一种基于信息熵表征isomiR表达鉴定乳腺癌生物标志物的方法
文献发布时间:2023-06-19 19:27:02
技术领域
本发明属于医药技术领域,具体涉及一种基于信息熵表征isomiR表达鉴定乳腺癌生物标志物的方法。
背景技术
乳腺癌(BRCA)是最常见的癌症之一,也是导致全球女性癌症死亡的主要原因,并且乳腺癌发病率呈现逐年升高的趋势。据世界卫生组织国际癌症研究机构(IARC)发布得2020年全球最新癌症数据,2020年全球新发226万例乳腺癌病例,首次超过肺癌(221万例)成为全球第一大癌症。筛选和预测与乳腺癌发生、发展和预后相关基因,以进行早期检测或治疗靶向可以提高乳腺癌患者的存活率,也为乳腺癌的研究提供新思路。
MicroRNA(miRNA)是一种长度约为22个核苷酸的小RNA分子,在以往研究中已证明miRNA在生命活动中参与各种各样的调节途径,发挥着重要的调控作用,可以通过与靶mRNA配对来控制基因表达水平。isomiR是miRNA位点产生的具有一系列具有序列和表达多样的多种异构体形式。主要源自于miRNA加工和成熟过程Drosha和Dicer的不精确和可选择性切割,pre-miRNA由Drosh处理的pri-miRNA产生,由5p臂,3p臂和末端环组成。随后,pre-miRNA通过输出蛋白5输出到细胞质中,在那里它们被Dicer切割以释放末端环和5p-3p双链体。最后,miR-5p或miR-3p臂被选择性地加载到RNA诱导沉默复合体(RISC)上,并靶向靶基因。
信息熵是生物信息学中的一个重要工具,在序列分析中有着广泛的应用。使用信息熵来表征isomiR表达,能够较无偏差得衡量isomiR在样本中表达的均衡与否,但将其应用于识别乳腺癌生物标志物方面的研究较少。
发明内容
目的:为了克服现有技术存在的不足,本发明提供一种基于信息熵表征isomiR表达鉴定乳腺癌生物标志物的方法,通过生物信息学方法在isomiR水平探索其在乳腺癌发生发展中的表达机制和生物学功能,并确定其作为乳腺癌生物标志物的潜在用途,对疾病的早期诊断或靶向治疗具有重要意义。
为达到上述目的,本发明是采用下述技术方案实现的:
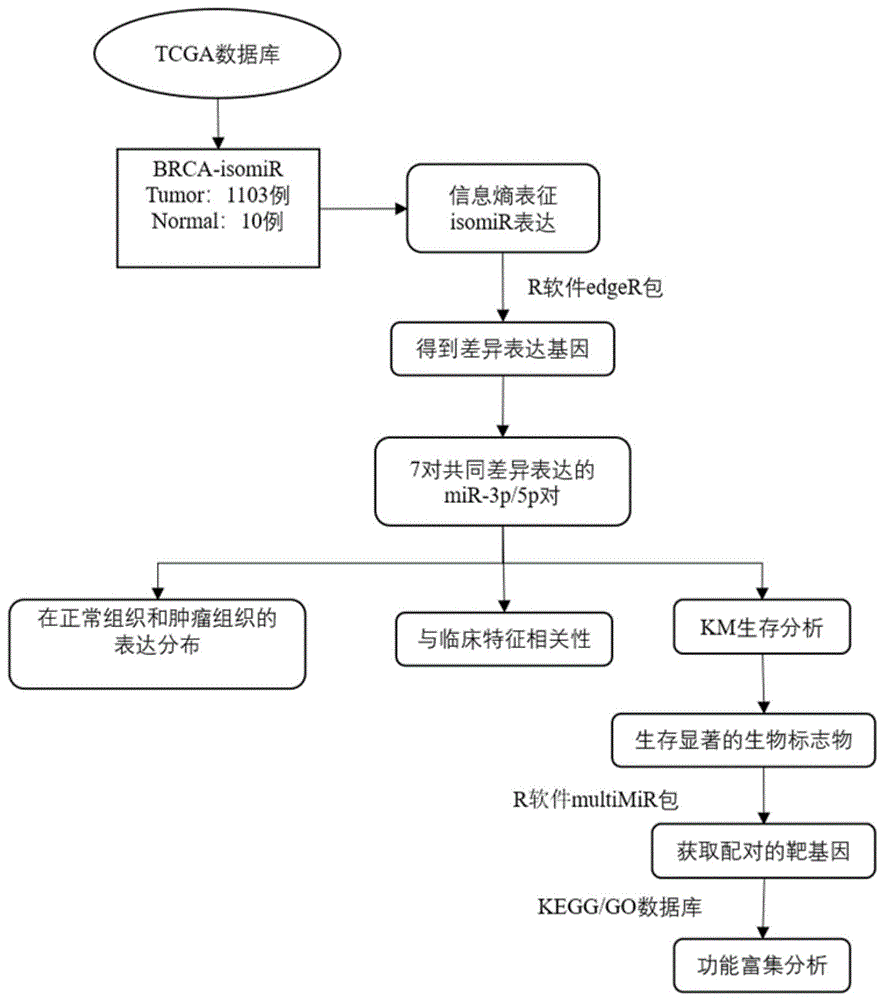
一种基于信息熵表征isomiR表达鉴定乳腺癌生物标志物的方法,其特征在于,包括以下步骤:
获取乳腺癌的isomiR表达数据和乳腺癌的临床信息;通过miRbase对isomiR表达数据进行注释,根据MIMAT号将pre-miRNA注释为5p/3p臂;分别基于pre-miRNA的两臂isomiR表达计算信息熵来表征pre-miRNA的表达,得到新的pre-miRNA的信息熵表达矩阵;
在得到的pre-miRNA信息熵表达数据中,挑选出两臂均优势表达的pre-miRNA,并计算出每个pre-miRNA的主要臂;
将均优势表达的pre-miRNA,以isomiR的最佳表达表征pre-miRNA在样本间的表达,使用信息熵来计算pre-miRNA在每个样本间的表达情况,得到由信息熵表征的表达矩阵,并进行差异表达分析,分析isomiR在乳腺癌肿瘤和癌旁组织中的表达水平,筛选出共同差异表达的miR-3p/5p对;
将miR-3p/5p对结合TCGA数据库的乳腺癌临床信息,分析miR-3p/5p对表达水平与各个临床变量之间的相关性;通过Kaplan-Meier分析miR-3p/5p对的表达水平与乳腺癌患者预后之间的相关性,预测可作为乳腺癌生物标志物的miR-3p/5p对。
进一步的,使用R软件miRBaseVersion.db包对isomiR的MIMAT号将pre-miRNA分别注释为5p/3p臂。
进一步的,基于两臂计算两臂的每个isomiR在每个pre-miRNA表达的信息熵,分别记为MIH
以两臂为基准,MIH
进一步的,对于两臂信息熵矩阵,分别计算两臂的平均信息熵,以此基准挑选出两臂均优势表达的pre-miRNA;并根据两臂的平均表达比,得出每个pre-miRNA的主要臂。
进一步的,对于isomiR在乳腺癌的表达,选取每个miRNA最佳表达的一条序列,并使用信息熵来表征miRNA在样本间的表达情况,得到行为miRNA,列为样本名的由信息熵表征的表达矩阵。
进一步的,信息熵计算公式如下:
H(x)为pre-miRNA在每个样本表达的信息熵,n为样本数目,i为当前样本号,P
进一步的,将由信息熵表征的表达矩阵使用R软件edgeR包进行差异表达分析,得到差异表达的miRNA;再筛选出共同差异表达的miR-3p/5p对,分析其在乳腺癌肿瘤组织和癌旁组织中的表达分布。
进一步的,差异基因表达分析的阈值条件均设置为:|log
进一步的,根据共同差异表达的miR-3p/5p对结合乳腺癌的临床信息进行分析,得到miR-3p/5p对的表达水平与临床变量之间的相关性。
进一步的,使用miR-3p/5p对的表达数据,通过R软件survival和survminer包,使用Kaplan-Meier方法,对上述miR-3p/5p对进行生存分析,以FDR<0.05作为筛选标准,得到可作为预测乳腺癌患者预后情况的生物标志物。
一种基于信息熵表征isomiR表达鉴定乳腺癌生物标志物的方法,包括以下步骤:
步骤1)下载数据。下载TCGA数据库中乳腺癌的isomiR表达数据和乳腺癌的临床信息。
步骤2)通过miRbase对isomiR进行注释,根据MIMAT号将pre-miRNA注释为5p/3p臂。分别基于pre-miRNA的两臂isomiR表达计算信息熵来表征pre-miRNA的表达,得到新的pre-miRNA的信息熵表达矩阵。
步骤3)对步骤2)得到的pre-miRNA信息熵表达数据,挑选出两臂均优势表达的pre-miRNA,并计算出每个pre-miRNA的主要臂。
步骤4)对步骤1)的数据,以isomiR的最佳表达表征pre-miRNA在样本间的表达,使用信息熵来计算pre-miRNA在每个样本间的表达情况,得到新的表达矩阵。
步骤5)对步骤4)的表达数据进行差异表达分析,分析isomiR在乳腺癌肿瘤和癌旁组织中的表达水平,并筛选出共同差异表达的miR-3p/5p对。
步骤6)根据步骤5)得到的miR-3p/5p对,结合TCGA数据库的乳腺癌临床信息,分析miR-3p/5p对表达水平与各个临床变量之间的相关性。
步骤7)通过Kaplan-Meier分析miR-3p/5p对的表达水平与乳腺癌患者预后之间的相关性,预测可作为乳腺癌生物标志物的miR-3p/5p对。
步骤8)根据步骤7)得到的生物标志物miR-3p/5p对,进行靶基因预测,提取其对应靶基因。
步骤9)基于靶基因,通过KEGG/GO途径进行功能分析,探索miR-3p/5p对调控靶基因的途径。
在一些实施例中,所述的基于信息熵表征isomiR表达鉴定乳腺癌生物标志物的方法,步骤1)所述的数据集包含TCGA数据库中乳腺癌isomiR的表达数据和TCGA数据库中乳腺癌的临床信息等。
在一些实施例中,所述的基于信息熵表征isomiR表达鉴定乳腺癌生物标志物的方法,步骤2)中,对isomiR进行注释,并基于两个臂的isomiR使用信息熵表征pre-miRNA的表达。包括:
使用R软件miRBaseVersion.db包对isomiR的MIMAT号将pre-miRNA分别注释为5p/3p臂。
分别基于pre-miRNA的两个臂,计算两个臂的每个isomiR在每个pre-miRNA表达的信息熵,得到行为pre-miRNA,列为5p、3p臂,值为信息熵的表达矩阵。
在一些实施例中,所述的基于信息熵表征isomiR表达鉴定乳腺癌生物标志物的方法,步骤3)中,对于两臂信息熵矩阵,分别计算两臂的平均信息熵,以此基准挑选出两臂均优势表达的pre-miRNA。并根据两臂的平均表达比,得出每个pre-miRNA的主要臂。
在一些实施例中,所述的基于信息熵表征isomiR表达鉴定乳腺癌生物标志物的方法,步骤4)中,对于isomiR在乳腺癌的表达,选取每个miRNA最佳表达的一条序列,并使用信息熵来表征miRNA在样本间的表达情况,得到行为miRNA,列为样本名的表达矩阵。
在一些实施例中,所述的基于信息熵表征isomiR表达鉴定乳腺癌生物标志物的方法,步骤5)中,根据步骤4)得到的表达矩阵,使用R软件edgeR包进行差异表达分析,得到差异表达的miRNA。再筛选出共同差异表达的miR-3p/5p对,分析其在正常组织和肿瘤组织的表达分布。
上述差异基因表达分析的阈值条件均设置为:|log
在一些实施例中,所述的基于信息熵表征isomiR表达鉴定乳腺癌生物标志物的方法,步骤6)中,根据步骤5)得出的差异miR-3p/5p对结合乳腺癌的临床信息进行分析,得到miR-3p/5p对的表达水平与临床变量之间的相关性。
在一些实施例中,所述的基于信息熵表征isomiR表达鉴定乳腺癌生物标志物的方法,步骤7)中,使用miR-3p/5p对的表达数据,通过R软件survival和survminer包,使用Kaplan-Meier方法,对上述miR-3p/5p对进行生存分析,以FDR<0.05作为筛选标准,进一步得到可作为预测乳腺癌患者预后情况的生物标志物。
在一些实施例中,所述的基于信息熵表征isomiR表达鉴定乳腺癌生物标志物的方法,步骤8)中,根据步骤7)得到的可能生物标志物,使用R软件multiMiR分别得到经过实验验证的生物标志物的靶向基因。
在一些实施例中,所述的基于信息熵表征isomiR表达鉴定乳腺癌生物标志物的方法,步骤9)中,根据步骤8)得到的靶向基因,分别通过KEGG/GO进行功能分析。探索miR-3p/5p调控靶基因的途径。
有益效果
本发明提供的基于信息熵表征isomiR表达鉴定乳腺癌生物标志物的方法,用客观、可量化的信息熵指标来评估isomiR的表达水平,可以更为准确地反应isomiR在样本间的表达差异性。能够提供有关乳腺癌的发生机制的信息,从而为乳腺癌的基因靶点治疗和筛选生物标志物提供新的思路和研究方向。
附图说明
图1是本发明实施例的方法流程图;
图2是本发明实施的一种基于信息熵表征isomiR表达鉴定乳腺癌生物标志物的方法的结果图。
具体实施方式
下面结合附图和实施例对本发明做进一步描述。以下实施例仅用于更加清楚地说明本发明的技术方案,而不能以此来限制本发明的保护范围。
实施例1
1.准备数据集。通过R软件TCGAbiolinks包从TCGA
(https://portal.gdc.cancer.gov)数据库中下载乳腺癌isomiR的染色体位置信息、isomiR在样本间的表达数据(样本包含癌组织1103例,正常组织104例)以及对应的年龄、肿瘤分期、TMN分期、生存时间、生存状态等临床信息。
2.对数据集进行分析,使用R软件miRBaseVersion.db包对isomiR的MIMAT号进行名称注释将pre-miRNA分别注释为5p/3p臂;基于两臂计算两臂的每个isomiR在每个pre-miRNA表达的信息熵,分别记为MIH
以两臂为基准,MIH
3.计算出两臂的平均表达信息熵,以其为阈值,挑选出两臂表达均高于平均值的pre-miRNA(n=324);并计算pre-miRNA两臂的表达比,得出主要臂。
实施例2
1.根据均优势表达的pre-miRNA在样本的表达情况,以其最佳表达的序列作为表征,使用表达值大于1000标准进行进一步筛选,得到256个miRNA。
2.计算在样本间表达的信息熵,得到由信息熵表征的表达矩阵。行名为基因名称(n=256),列名为样本名称(n=1207)。其信息熵计算公式如下:
H(x)为pre-miRNA在每个样本表达的信息熵,n为样本数目,i为当前样本号,P
3.根据上述的表达矩阵,以正常组织为对照,使用R软件edgeR包进行差异表达分析,筛选标准为|log
4.结合乳腺癌的临床信息进行分析,得到miR-3p/5p对的表达水平和临床变量之间的相关性(表2);多数基因的高表达与患者更晚的T分期、M分期、N分期显著相关,表明基因的高表达水平可能会促使乳腺癌浸润范围的增加,使肿瘤转移风险升高,导致患者患病恶性程度的增加。
5.通过R软件survival和survminer包,使用Kaplan-Meier方法进行生存分析,以FDR<0.05作为筛选标准,最终得到可作为预测乳腺癌患者预后情况的生物标志物(miR-767-5p/3p对、miR-877-5p/3p对)。高表达的5p臂和低表达的3p臂与较差的生存率相关。
6.根据得到的生物标志物,通过R软件multiMiR获取其配对的经过实验验证的靶基因,并进行功能富集分析。发现两对生物标志物介导的miRNA靶标网络在乳腺癌发生中起重要作用。miR-767-5p靶基因主要富集在泛素介导的蛋白水解,病毒致癌作用等通路;miR-767-3p靶基因主要富集在病毒致癌作用,氨基酸和核苷酸糖代谢,核质运输等通路;miR-877-5p靶基因主要富集在细胞衰老,神经营养素信号通路,PI3K信号通路;miR-877-3p靶基因主要富集在核质运输,HIF-1信号通路。miRNA与其对应靶基因可能通过上述通路进行调控,参与乳腺癌的发生发展。
7.miRNA的臂稳态被破坏时,miRNA功能发生变化,可能导致疾病进展。在乳腺癌中,miR-767和miR-877的臂偏好可能是乳腺癌发生和进展的关键介质,其表达可作为预测乳腺癌的预后指标。重新修饰miR-767以及miR-877的靶点稳态可能代表乳腺癌预防和治疗的现实方法。
表1:edgeR算法得到的共同差异差异表达的miR-3p/5p对
表2:miR-3p/5p对表达与临床病理学特征之间的关系
/>