一种基于原子特征传递网络的小分子单步逆合成预测方法
文献发布时间:2023-06-19 19:28:50
技术领域
本发明属于计算机辅助药物研发技术领域,具体涉及一种基于原子特征传递网络的小分子单步逆合成预测方法。
背景技术
治疗特定疾病药物的研发,从最初的实验室研究、临床试验到最终上市,都是一个高投资、高风险、长周期的项目。现代药物开发旨在通过在药物发现阶段和临床前阶段使用机器学习技术,如靶标识别和验证、虚拟筛选、先导物优化等,加快中间过程和降低成本。尽管在过去的几十年里取得了重大进展,但有机合成仍然是药物发现的一个难题。在早期,一直由化学领域的专家依靠自身资深的经验完成这个任务,这对于他们的背景要求是非常高的,而且受制于人脑算力不足,依靠专家经验推荐一条合成路径最少需要3h。逆合成计划的目的是将目标分子转化为更容易获得的前体,找到有效的合成途径。
近年来,计算机辅助合成规划(computer-assisted synthetic planning,CASP)的快速发展,特别是逆合成预测得到了广泛关注,它为目标分子每设计对应的合成路径仅仅需要5-10分钟;而且它还可以同时推荐出多条需要不同底物的合成路线,研究人员可以根据自己的实验条件和需求进行特异性选择。不过当前这些CASP工具存在一个最主要的限制就是,它们所应用的单步逆合成预测策略都是基于模板的预测方法,这种范式思路的做法是将化学反应的规则编码到了计算机中,缺少泛化性,无法对模板之外的药物分子进行逆合成路径的预测,并且,随着新知识的发现,这些模板需要经常更新,这也是一项非常繁琐的工作。因此,无模板单步逆合成预测算法的研究和开发对未来药物研发领域更为重要,本发明的主要目的是开发一个无模板的单步逆合成预测模型,为未来药物逆合成路线的设计提供帮助。
与正向反应预测相反,逆合成是一种从产物分子到廉价和可获得的反应物的反向外推。逆合成分析可以有效地解决复杂分子的合成问题,促进有机合成科学的发展。此外,随着系统生物学实验技术的进步和实验数据的不断积累,大量的生物医学数据已经出现,并为数据划分合理的生物合成设计提供了动力。深度学习(DL)是人工智能机器学习(ML)的一个子领域,它可以直接从原始数据中理解和学习其内在规律和复杂表示。因此,应用深度学习(DL)的新尝试逐渐进入舞台,为化学合成研究开辟了新的范式。
随着机器翻译的发展越来越受到人们的关注,为无模板提供可能,一些研究人员发现机器翻译和逆合成之间的类比是明显的。目前,大多数无模板单步逆合成任务的研究都基于LSTM、Trasformer等seq2seq算法(以及它们的变体)开发了分子翻译的模型,这类方法忽视了一个重要的问题,那就是药物分子本身做为一个图,它包含丰富了结构信息。因此,许多基于GNN的研究已经出现在了逆合成技术上。它可以通过递归传递分子图的信息来聚合每个原子的表示来学习每个原子的表示。不过它们这类以生成为基础的模型也是存在一个比较明显的不足,那就是缺少了对原子本身属性以及周围原子所带来影响的考虑。然而分子之间能发生化学反应恰恰是因为一些关键的原子起到了足够重要的作用。
在有机合成领域中,一条唯一且关键的知识就是寻找目标分子中容易断裂的化学键,另一方面,键能是从能量因素衡量化学键强弱的物理量,因此在区别可断裂化学键与其他键的时候,这也是一条重要的需要考虑的指标,不能在设计模型的时候把它忽略掉。基于以上两点,以及其他研究中的不足,本申请设计了一个基于原子特征传递网络与对比学习的深度学习模型RetroAFPNN,用于分析目标分子中易断裂的化学键,进而完成它的单步逆合成预测。
发明内容
本发明提供了一种基于原子特征传递网络的小分子单步逆合成预测方法,采用一个无模板的单步逆合成模型RetroAFPNN,解决了一般逆合成工具中不能对模板之外的分子进行预测的问题。并且相对于生成类的模型,本发明考虑到其对原子关注度不足的问题,达到了一个更高准确率的表现。
为实现上述目的,本发明所提供的技术解决方案是:
一种基于原子特征传递网络的小分子单步逆合成预测方法,其特殊之处在于,包括以下步骤:
1)利用目标分子断裂位点识别模型对断裂位点进行预测
1.1)构建目标分子断裂位点识别模型
所述目标分子断裂位点识别模型包括两原子特征传递网络层以及一个全连接层;
1.2)训练步骤1.1)构建的目标分子断裂位点识别模型
1.2.1)数据采集
采集目标分子断裂位点识别模型训练与测试过程中所需的化学反应数据,并将其按比例划分为训练集和测试集;
1.2.2)数据处理
将步骤1.2.1)得到的所有化学反应数据处理为Smiles类型数据;
1.2.3)构建原子的初始特征
针对步骤1.2.2)得到的数据中的每个化学分子,构建分子中每个原子的初始特征;
1.2.4)利用两层原子特征传递网络层(Atomic Feature Passing NeuralNetwork,AFPNN)重构1.2.3)得到的原子的初始特征
构建目标分子的拓扑结构图,通过两层原子特征传递网络层(Atomic FeaturePassing Neural Network,AFPNN),聚合每个原子周围与它有连边的其它原子之间的特征来重构该原子的特征,得到该原子重构后的特征;这里AFPNN主要的功能是聚合每个原子周围和它有连边的其他原子之间的特征,以此来重构该原子的特征。
1.2.5)构建键特征
通过加和每个键两端原子重构后的特征,来构建所有键的特征,每个键形成一个样本,最终,得到所有分子中所有样本的特征,并对样本标注正负标签y;这里指的是,针对每一个分子中的每一个化学键,都采用加和其两端原子特征的方法构建这个键的特征,每个键都是一个样本,有的是正样本,有的是负样本,其判断依据是看该化学键是否是断裂键,如果是的话,为正样本,否则为负样本。
1.2.6)通过全连接层模型将键特征映射到一维空间
利用全连接层(FC)将步骤1.2.5)构建的键特征映射到1维,得到所有键特征映射到1维之后的特征结果
1.2.7)负反馈调节
利用交叉熵损失函数计算步骤1.2.6)得到的特征结果
模型训练时,需要不停的进行重复,以更新模型中的参数,从而使得训练集中对化学键预测得到的标签与它真实的标签之间的差距最小,才能完成模型训练。
1.3)利用步骤1.2)训练好的目标分子断裂位点识别模型对目标分子的断裂位点进行预测;
2)利用合成子到反应物的转换模型SR-FC,推荐对应的反应物
2.1)针对目标分子,以步骤1)预测的断裂键为中心,获得拓扑深度为s的子结构作为代表该目标分子核心结构;
2.2)通过Rdkit中的函数将目标分子在正确的断裂位置断裂,形成合成子;
2.3)将步骤2.2)得到的合成子与其所对应的反应物进行比较,统计两者之间的差异结构,构建合成子到反应物转换时所需要添加额外基团的数据库;
2.4)将步骤2.3)得到的额外基团两两组合,并进行One-Hot编码,组成多组标签;
2.5)通过MACCSkeys提取步骤2.1)所得目标分子核心结构的分子指纹特征,再通过两层全连接层构建其与步骤2.4)所得标签之间的函数映射关系,经过迭代训练,得到合成子到反应物的转换模型SR-FC;
2.6)利用步骤2.5)得到的合成子到反应物的转换模型SR-FC,推荐对应的反应物,完成逆合成预测。
进一步地,步骤1.2.1)中,所述化学反应数据均从美国USPTO中采集,其中包含50K条化学反应数据。
进一步地,步骤1.2.2)中,采用Rdkit中对化学反应读取的算法来整理步骤1.2.1)采集到的化学反应数据,将所有化学反应数据处理成统一标准的Smiles类型数据。
进一步地,步骤1.2.3)中原子初始特征构建的方法具体如下:
针对化学分子中的每个原子,提取以下特征,并将所有特征拼接成特征向量;
①采用One-Hot编码方式表示每个原子的种类,特征长度23维;
②计算每个原子的度,特征长度1维;
③判断原子是否属于芳香环,用0、1表示,,特征长度1维;
④计算与该原子连接的氢原子的个数,特征长度1维;
⑤计算该原子所带的电荷数,特征长度1维;
⑥统计该原子的原子质量,特征长度1维;
拼接后每个原子的特征向量长度为以上特征的长度之和28维。
进一步地,步骤1.2.4)重构的具体方法如下:
A1.数学建模
步骤1.2.3)中构建了目标分子中原子的原始特征,特征向量长度为28维;其中,第i个原子用A
目标分子D={A
采用e
A2.归一化
为了避免数据特征的淹没,对所构建原子的原始特征进行归一化处理:
其中,n是目标分子中的原子总数;
A3.原子特征传递网络层的构建
构建原子特征传递网络层以更新目标分子D中所有原子的原始特征,
其中,tanh是激活函数;
(·,·)表示连接关系;
W
b表示是可训练的偏置;
N(i)是在目标分子D中原子A
使用两层的原子特征传递网络层来更新原子特征,更新后的原子
其中,relu是激活函数。
进一步地,步骤1.2.5)中构建键特征的具体方法如下:
在目标分子D中,键K
其中,
表示分子D中第j个原子重构后的特征;
表示向量的拼接;
W
e
进一步地,步骤1.2.7)中采用的交叉熵损失函数为:
/>
其中,m为样本的数量。
进一步地,步骤2.1)中,拓扑深度S取1;
步骤2.5)中,采用Rdkit将核心结构的特征利用二进制表示,MDL公司开发的MACCSkeys指纹共拥有166个特征,但是MACCSkeys总长度为167bits,第0位为占位符,第1-166位为分子特征位;用该方法提取目标分子断裂键周围拓扑深度为1的核心结构的分子指纹,每个分子指纹长度为167bits。
同时,本发明还提供了一种计算机可读存储介质,其上存储有计算机程序,其特殊之处在于:所述计算机程序被处理器执行时实现上述方法的步骤。
一种电子设备,其特殊之处在于:包括处理器和计算机可读存储介质;所述计算机可读存储介质上存储有计算机程序,所述计算机程序被所述处理器运行时执行上述方法的步骤。
本发明的原理:
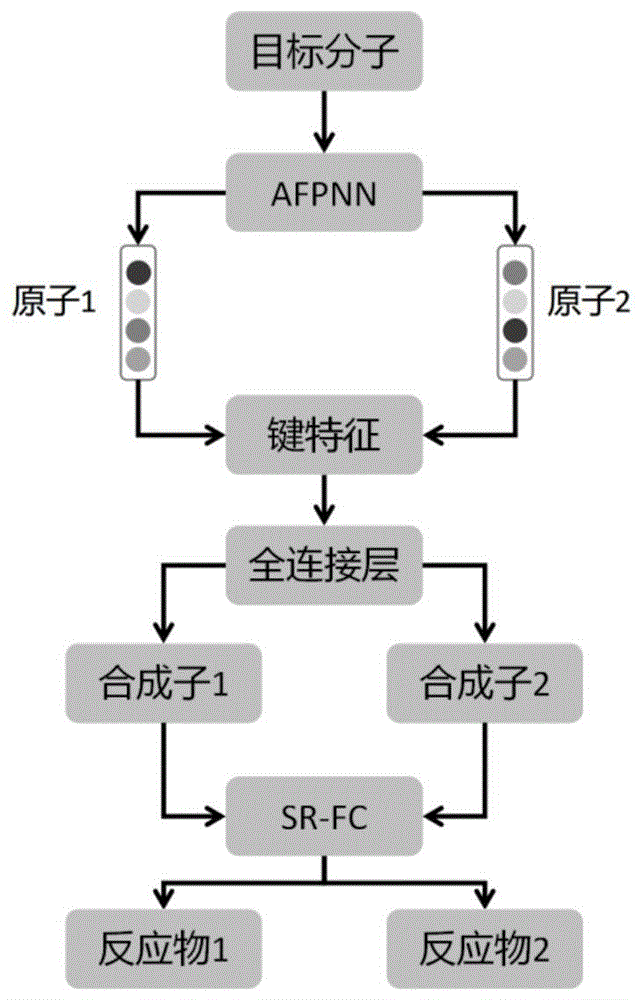
本发明利用原子特征传递网络AFPNN,设计了一种单步逆合成预测模型RetroAFPNN,该模型可以对复杂的药物分子进行逆合成的推断。第一部分目标分子断裂位点的识别,利用原子特征传递网络AFPNN对目标分子中原子的特征进行重构后,再进行对键的特征的构建,最终通过一个全连接层,输出最终对断裂位点的预测结果。第二部分反应物的推荐,构建目标分子断裂键周围拓扑深度为n的核心结构与反应物之间的映射关系,在目标分子断裂为合成子之后,推荐对应的反应物。
本发明的优点:
本发明主要采用了原子特征聚合网络AFPNN捕捉原子的特征,以及使用键能作为它们之间的权重扰动,接着,用于识别目标分子中最易断裂的化学键,最后构建了合成子到反应物的转换模型SR-FC,完成目标分子的单步逆合成预测。本发明在机器学习中融入专业领域知识,深度的从研究人员完成逆合成工作的思路去考虑了逆合成中的规律,预测时间短,准确率更高。
附图说明
图1是本发明整体的预测流程图;
图2是本发明中对于目标分子断裂位点识别模型的构建流程图;
图3是本发明中对于反应物推荐模型SR-FC的构建流程图。
具体实施方式
以下结合附图和具体实施例对本发明的内容作进一步的详细描述:
本发明根据原子特征传递网络的特性,提出了一种基于原子特征传递网络的小分子单步逆合成预测方法。本方法分为两个阶段,第一阶段是对目标分子断裂位点的识别阶段,第二阶段是对反应物的推断阶段。本发明经过测试具有较高的准确性,为后续逆合成预测工作的发展提供了一定的基础。
依据本发明提出的一种基于原子特征传递网络的小分子单步逆合成预测方法的一个实施例具体如下:
本发明所提出的模型训练使用化学反应数据集USPTO-50K,该数据集中包含50000条化学反应的数据。按照9∶1,将数据集分为训练集和测试集。
本发明可用于小分子单步逆合成的预测,针对原始数据,本发明采用Rdkit中对化学反应读取的算法来整理所采集到的化学反应数据,将所有化学反应数据处理成统一标准的Smiles类型数据,接着再根据目标分子中原子之间的拓扑关系,将化合物转换为图数据。同时,构建每个原子的原始特征。
针对每一个分子的图数据,结合其原子的原始特征,采用原子特征传递网络层AFPNN对原子的初始特征进行重构。
利用重构后的原子,采用特征加和的方法,构建对应键的特征,同时标注正负样本的标签。
接着,采用全连接层对键的特征进行映射,得到计算结果。
最后,采用交叉熵损失函数计算结果与标签之间的损失,并根据损失残差通过负反馈调节来训练模型中的参数。
训练完成后,就得到了一个基于原子特征传递网络的小分子断裂位点识别模型。
为了评价模型的性能,本发明采用五折交叉验证的方法在测试集上计算了断裂位点识别的准确率和反应物推荐的准确率,测试结果如表1所示:
表1断裂位点识别模型性能展示
随后,对训练集中目标分子中以断裂键为中心拓扑深度为1的子结构进行统计,并统计合成子与反应物之间的差别,然后,采用两层全连接层构建核心子结构到额外基团的映射函数,经过训练,得到了合成子到反应物的转换模型SR-FC。
为了评价模型的性能,本发明在测试集数据上测试了模型的准确率,经过测试,SR-FC对于目标分子核心子结构到反应物映射的识别率达到了0.895。
最后,本发明在测试集上统计了将“断裂位点识别模型”与“合成子到反应物的转换模型”结合在一起的准确率,并且在同一标准下与其它比较先进的模型进行了比较,本发明方法开发的模型表现出了优异的性能。结果如表2所示:
表2小分子单步逆合成综合模型RetroAFPNN性能展示
从测试结果中可以看出,本发明对于小分子单步逆合成路径的预测准确率较高。其中,Top5的推荐准确率达到了0.880,具有显著的效果。
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明公开的技术范围内,可轻易想到各种等效的修改或替换,这些修改或替换都应涵盖在本发明的保护范围之内。