一种临床病原微生物宏基因组完整性检测方法
文献发布时间:2024-04-18 19:53:33
技术领域
本申请属于基因检测领域,具体涉及一种临床病原微生物宏基因组完整性检测方法。
背景技术
宏基因组测序(mNGS)在感染性疾病诊断中发挥着重要作用,依靠生物信息学手段对mNGS微生物进行筛选、过滤、比对、物种注释等系统的生物信息学分析,对临床感染疾病的精准诊疗具有重要的指导意义。在分子生物学实验中,基因完整性是极重要的质控指标之一,例如,全基因组测序、全外显子组测序和目标区域扩增子测序等,都要求基因完整,否则实验会面临结果变差甚至失败的风险。然而,现有的基因完整性检测方法普遍存在成本高、效率低等问题。
发明内容
为了解决上述技术问题,本申请提供了一种临床病原微生物宏基因组完整性检测方法。
包括以下步骤:
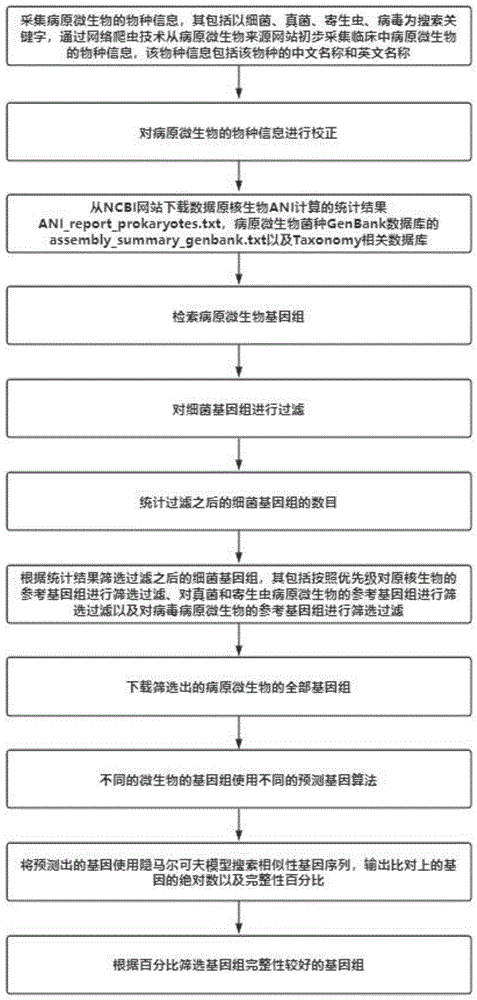
采集病原微生物的物种信息,其包括以细菌、真菌、寄生虫、病毒为搜索关键字,通过网络爬虫技术从病原微生物来源网站初步采集临床中病原微生物的物种信息,该物种信息包括该物种的中文名称和英文名称;
对病原微生物的物种信息进行校正;
从NCBI网站下载数据原核生物ANI计算的统计结果ANI_report_prokaryotes.txt,病原微生物菌种GenBank数据库的assembly_summary_genbank.txt以及Taxonomy相关数据库;
检索病原微生物基因组;
对细菌基因组进行过滤;
统计过滤之后的细菌基因组的数目;
根据统计结果筛选过滤之后的细菌基因组,其包括按照优先级对原核生物的参考基因组进行筛选过滤、对真菌和寄生虫病原微生物的参考基因组进行筛选过滤以及对病毒病原微生物的参考基因组进行筛选过滤;
下载筛选出的病原微生物的全部基因组;
不同的微生物的基因组使用不同的预测基因算法;
将预测出的基因使用隐马尔可夫模型搜索相似性基因序列,输出比对上的基因的绝对数以及完整性百分比;
根据百分比筛选基因组完整性较好的基因组。
优选的,所述对病原微生物的物种信息进行校正的过程为:
进一步的,所述检索病原微生物基因组的过程为:
以校正病原微生物信息后的种水平的物种分类编号为关键字,使用python字段匹配的原理从GenBank数据库中检索获得的物种分类编号对应的所有参考基因组。
进一步的,所述对细菌基因组进行过滤的过程为:
将从GenBank数据库检索得到的包含全部基因组相关信息的数据框中的″assembly_accession″与NCBI中对所有原核生物ANI计算的统计结果ANI_report_prokaryotes.txt中的″genbank-accession"作为关键字,使用python中的字段匹配函数merge()对assembly_summary_genbank.txt文件和ANI_report_prokaryotes.txt文件进行连接,并使用python语法构建比较运算方法,以保留ANI计算结果与声明的物种一致且最佳匹配的物种与声明的物种一致的基因组。
进一步的,所述统计过滤之后的细菌基因组的数目包括统计每一个物种的全部基因组的数目以及各物种中四个组装水平下基因组的数目,具体为:
以物种分类编号为关键字,使用python中的value_counts()统计关键字出现的次数,从而得到每一个物种的全部基因组的数目;
采用python语法中的参数by设定标签,参数level设定索引的位置并进行分组,提取统计的关键字,再根据关键字使用词频统计函数value_counts()统计该物种中四个组装水平下基因组的数目。
进一步的,所述按照优先级对原核生物的参考基因组进行筛选过滤的过程为:
采用python语法构建比较运算符,以用于判断在NCBI的RefSeq项目分类中参考基因组是否为Reference genome或representative genome,如果是Reference genome或representative genome,则保留该参考基因组;
反之,则采用python语法构建比较运算,以用于判断统计得到的每一个物种的全部基因组的数目;
如果某个病原微生物的全部基因组的数目小于或等于预设条数,则保留全部基因组作为该物种的基因组;
如果该病原微生物下的全部基因组的数目大于预设条数,则取该病原微生物全部基因组数目的5%~15%作为该物种的基因组。
进一步的,所述下载筛选出的病原微生物的全部基因组的过程为:
采用字段匹配方法将筛选出来的全部基因组与病原微生物菌种GenBank数据库的assembly_summary_genbank.txt中的″assembly_accession″列匹配并获取每一个基因组的下载地址,最后得到每一个基因组对应的下载地址列表;
以下载地址列表为输入文件,编写python脚本自动下载基因组;对下载后的基因组进行md5校验,确保文件传输无异常后解压。
进一步的,不同的微生物的基因组使用不同的预测基因算法;针对细菌和病毒的微生物的基因组,采用prodigal的预测基因的算法,针对真核生物,采用metaeuk的预测基因的算法。
进一步的,所述输出比对上的基因的绝对数以及完整性百分比,评估结果包括:
C:多少个基因被覆盖以及覆盖基因占总基因数的百分比,计算公式分别为C=S+D,C%=C/Toal*100%;其中,C为Complete;
S:多少个基因经过比对发现是单拷贝以及单拷贝基因占总基因数的百分比;S%=S/Toal*100%;其中,S为Complete and single-copy;
D:多少个基因经过比对发现包含多拷贝以及多拷贝基因占总基因数的百分比;D%=D/Toal*100%;其中,D为Complete and duplicated;
F:多少个基因经过比对覆盖不完全,只是部分比对上以及部分比对上的基因占总基因数的百分比;F%=F/Toal*100%;其中,F为CFragmented;
M:没有比对结果成功的基因数以及错配基因数占总基因数的百分比;M%=M/Toal*100%;其中,C为Missing;
Total:总共测试的基因条目数,Total=C+F+M;其中,Total为Total groupssearched。
进一步的,根据百分比筛选基因组完整性较好的基因组包括根据C百分比筛选基因组完整性较好的基因组其判断方法为,预先设定完整性满足预设需求的经验值作为阈值;当同一个物种拥有多条基因组时,保留同一个物种的完整性大于等于阈值的基因组,去除完整性不足的基因组;
筛选得到的基因组需要满足以下条件:
种类上:所筛选得到的基因组至少在种及以下水平;
基因组长度:所得到的基因组其组装水平在scaffold及以上,将Scaffolds按照长度排序,由长到短进行加和,总长度达到或者超过Scaffolds总长50%;
基因数:基因组中的基因占该谱系上的物种本身基因总数的大部分。
本申请的上述技术方案,相比现有技术具有以下技术效果:
本申请从国际公共数据库中收集临床中重要的病原微生物的基因组,基于距离矩阵建立系统发育进化树修正标记错误的序列,基于核心基因评估基因组序列的完整性和污染物区域的方式获取到高质量基因组,采用基于marker genes评估基因组序列的方法评估基因组的完整性,能够大大降低检测成本,提高检测效率。
应了解的是,上述一般描述及以下具体实施方式仅为示例性及阐释性的,其并不能限制本申请所欲主张的范围。
附图说明
为了更清楚地说明本申请实施例中的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本申请的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其它的附图。
图1为本申请具体实施方式提供的一种临床病原微生物宏基因组完整性检测方法的流程图。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
如图1所示,本申请提供的一种临床病原微生物宏基因组完整性检测方法包括以下步骤:
步骤100采集病原微生物的物种信息;
以细菌、真菌、寄生虫、病毒为搜索关键字,通过多种网络爬虫技术从CHIFNET中国医院侵袭性真菌病监测网、ABX指南、Uptodate网站、人间传染的病原微生物名录等病原微生物来源网站初步采集临床中重要的病原微生物的物种信息,该物种信息包括该物种的中文名称和英文名称。
例如,具体到种水平的重要病原菌可以为金黄色葡萄球菌(Staphylococcusaureus)、铜绿假单胞菌(Pseudomonas aeruginosa)、烟曲霉(Aspergillus fumigata)、白念珠菌(Candida albicans)、微小隐孢子虫(Cryptosporidium microscopices)、恶性疟原虫(Plasmodium falciparum)、猴痘病毒(Monkeypox Virus)和人疱疹病毒(Human herpesvirus)等。
步骤200对病原微生物的物种信息进行校正;
由于步骤100采集到的病原微生物的名称存在名称变动,格式复杂,病原物种层级不整齐等问题,因此需要对病原微生物的物种信息进行校正。
将步骤100采集到的病原微生物的名称作为关键字,采用聚焦网络爬虫技术在主流文献、词典和权威数据库(主要NCBI中的Taxonomy数据库)中进行检索,得到该病原微生物的名称对应的物种分类编号。
以物种分类编号为关键字对采集到的物种名称是否为物种的科学名称进行校对。
根据物种分类编号和物种的科学名称确定物种的分类学水平,保留在种水平的物种信息。其中,保留的在种水平的物种信息包括物种的科学名称、物种的分类学水平以及病原微生物的名称对应的物种分类编号(species-taxid)。
步骤300获取国际权威的公共数据库中的统计信息;
使用Aspera(ascp命令)从NCBI(National Center for BiotechnologyInformation,美国国家生物信息中心)网站高速下载数据原核生物ANI(AverageNucleotide Identity,平均核苷酸序列相似性)计算的统计结果ANI_report_prokaryotes.txt,病原微生物菌种GenBank数据库的assembly_summary_genbank.txt以及Taxonomy相关数据库。
步骤400检索病原微生物基因组;
以校正病原微生物信息后的种水平的物种分类编号为关键字,使用python字段匹配的原理从GenBank数据库中检索步骤S2中获得的物种分类编号对应的所有参考基因组。
步骤500对细菌基因组进行过滤;
从GenBank数据库检索得到的全部基因组的相关信息保存在包含多列的assembly_summary_genbank.txt文件中,NCBl中对所有原核生物ANI计算的统计结果保存在包含多列的ANI_report_prokaryotes.txt文件中。
将步骤400中从GenBank数据库检索得到的包含全部基因组相关信息的数据框中的″assembly_accession(组装编号)″与NCBI中对所有原核生物ANI计算的统计结果ANI_report_prokaryotes.txt中的″genbank-accession(genbank编号)″作为关键字,使用python中的字段匹配函数merge()对assembly_summary_genbank.txt文件和ANI_report_prokaryotes.txt文件进行连接,并使用python语法构建比较运算方法,以保留ANI计算结果与声明的物种一致且最佳匹配的物种与声明的物种一致的基因组。
步骤600统计过滤之后的细菌基因组的数目;
其包括统计每一个物种的全部基因组的数目以及各物种中四个组装水平下基因组的数目,具体为:
以物种分类编号为关键字,使用python中的value_counts()统计关键字出现的次数,从而得到每一个物种的全部基因组的数目。
统计每一个物种各组装水平下的数目,采用python语法中的参数by设定标签,参数level设定索引的位置并进行分组,提取统计的关键字(每一个物种的四个组装水平),再根据关键字使用词频统计函数value_counts()统计该物种中四个组装水平下基因组的数目。
步骤700根据统计结果筛选过滤之后的细菌基因组,其包括按照优先级对原核生物的参考基因组进行筛选过滤、对真菌和寄生虫病原微生物的参考基因组进行筛选过滤以及对病毒病原微生物的参考基因组进行筛选过滤。
具体地,按照优先级对原核生物的参考基因组进行筛选过滤的过程为:
①采用python语法构建比较运算符,以用于判断在NCBI的RefSeq项目分类中参考基因组是否为Reference genome或representative genome,如果是Reference genome或representative genome,则保留该参考基因组,反之,则进行步骤②;
②采用python语法构建比较运算,以用于判断步骤S6中统计得到的每一个物种的全部基因组的数目;
如果某个病原微生物的全部基因组的数目小于或等于预设条数,则保留该物种的全部基因组作为本次构建数据库时该物种的基因组;
如果该病原微生物下的全部基因组的数目大于预设条数,则取该病原微生物全部基因组数目的5%~15%作为该物种在本次构建数据库中该物种的基因组。
全部基因组数目的5%~15%通过以下方式获得:
按照该参考基因组是否为Reference genome、representative genome,基因组组装水平为Complete Genome水平、Chromosome水平、Scaffold水平、Contig水平的优先级从高到低筛选过滤基因组。
参考基因组的信息存储在一个包含多列的数据表中,例如,第一列为参考基因组的accession号;第二列为RefSeq项目分类信息(包含Reference genome,representativegenome或者不分类的信息,一条基因组只能是其中一种情况);第三列为组装水平(Complete Genome或Chromosome或Scaffold或Contig,一条基因组只能是其中一种情况)。
假设一个物种有100条基因组,预设条数为50。这个物种一共100条基因组已经超过预设条数50,因此,将筛选10条基因组代表该物种的基因组。如:其中一条基因组为GCA_000002135.3,在第二列中RefSeq项目分类信息为Reference genome或者representativegenome,则直接保留基因组,继续根据第三列的组装水平筛选剩余的9条基因组,如果在第二列中RefSeq项目无分类信息,则根据第三列筛选10条基因组。假设该物种CompleteGenome水平的有2条、Chromosome水平的有3条、Scaffold水平的3条、Contig水平92条),则这所选的10条基因组:由2条Complete Genome水平、3条Chromosome水平、3条Scaffold水平,2条contig水平组成。
具体地,对真菌和寄生虫病原微生物的参考基因组进行筛选过滤的过程为:
采用python语法构建比较运算符,以用于判断在NCBI的RefSeq项目分类中该参考基因组是否为Reference genome或representative genome,如果是Reference genome或representative genome,则保留该参考基因组,反之,则过滤;
具体地,对病毒病原微生物的参考基因组进行筛选过滤的过程为:
采用python判断病原微生物的种类,如果该病原微生物是病毒,则保留所有该病毒的所有参考基因组。
步骤800下载筛选出的病原微生物的全部基因组;
采用字段匹配方法将筛选出来的全部基因组与病原微生物菌种GenBank数据库的assembly_summary_genbank.txt中的″assembly_accession"列匹配并获取每一个基因组的下载地址(ftp),最后得到每一个基因组对应的下载地址列表;再以下载地址列表为输入文件,编写python脚本自动下载基因组;对下载后的基因组进行md5校验,确保文件传输无异常后解压。
步骤900不同的微生物的基因组使用不同的预测基因算法
针对细菌和病毒的微生物的基因组,采用prodigal的预测基因的算法预测基因组中的可能存在的基因;针对真核生物,采用metaeuk的预测基因的算法预测真核生物基因组中可能存在的基因;
步骤1000将预测出的基因使用隐马尔可夫模型搜索相似性基因序列,输出比对上的基因的绝对数以及完整性百分比;
将预测出的基因使用隐马尔可夫模型(hmmsearch)搜索相似性基因序列,根据计算过程中的得分判断单拷贝基因是否存在,根据该单拷贝基因家族的长度分布,预测基因出的基因长度必须落在平均长度的两个标准差(±2σ)之间即为完整;
步骤1100根据百分比筛选基因组完整性较好的基因组;
输出比对上的基因的绝对数以及完整性百分比,评估结果包括:
C(Complete):多少个基因被覆盖以及覆盖基因占总基因数的百分比,计算公式分别为C=S+D,C%=C/Toal*100%,;
S(Complete and single-copy):多少个基因经过比对发现是单拷贝以及单拷贝基因占总基因数的百分比;S%=S/Toal*100%
D(Complete and duplicated):多少个基因经过比对发现包含多拷贝以及多拷贝基因占总基因数的百分比;D%=D/Toal*100%
F(Fragmented):多少个基因经过比对覆盖不完全,只是部分比对上以及部分比对上的基因占总基因数的百分比;F%=F/Toal*100%
M(Missing):没有比对结果成功的基因数以及错配基因数占总基因数的百分比;M%=M/Toal*100%
Total(Total groups searched):总共测试的基因条目数,Total=C+F+M。
根据C(Complete)百分比筛选基因组完整性较好的基因组其判断方法为,人为预先设定完整性满足预设需求的经验值作为阈值。当同一个物种拥有多条基因组时,保留同一个物种的完整性大于等于阈值的基因组,去除完整性不足的基因组;
筛选得到的基因组需要满足以下条件:
1.种类上:所筛选得到的基因组至少在种及以下水平;
2.基因组长度:所得到的基因组其组装水平在scaffold及以上,将Scaffolds按照长度排序,由长到短进行加和,总长度达到或者超过Scaffolds总长50%;
3.基因数:基因组中的基因占该谱系上的物种本身基因总数的大部分。
当该物种的全部基因组的完整性都小于经验值,则将该物种内的基因组完整性从高到低进行排序,最终选择完整性满足预设需求的基因组;
当某些物种的基因组只有一条并且完整性低于经验值,则无论基因组的完整度为多少,都保留该基因组。
需要注意的是,上述仅为本发明的较佳实施例及所运用技术原理。本领域技术人员会理解,本发明不限于这里所述的特定实施例,对本领域技术人员来说能够进行各种明显的变化、重新调整和替代而不会脱离本发明的保护范围。因此,虽然通过以上实施例对本发明进行了较为详细的说明,但是本发明不仅仅限于以上实施例,在不脱离本发明构思的情况下,还可以包括更多其它等效实施例,而本发明的范围由所附的权利要求范围决定。