一种迭代分析生物学大样本数据的知识推理方法及系统
文献发布时间:2024-04-18 19:53:33
技术领域
本发明属于基于特定计算模型的计算机系统领域,尤其涉及一种迭代分析生物学大样本数据的知识推理方法、系统及装置。
背景技术
知识推理通过各种方法获取新的结论。其过程是在已有知识的基础之上,推断出未知的知识,通过从已知的知识出发,通过已经获取的知识,从中获取到所蕴含的新的事实,或者从大量的已有的知识中进行归纳,从个体知识推广到一般性的知识。对于知识推理而言,其包括的内容可以分为两种,第一种是已经知道的,用于进行推理的已有知识,另外一种是运用现有的知识推导或者归纳出来的新的知识。对于知识而言,其形式是多种多样的,可以是一个或者多个段落描述,又或者如传统的三段论的形式。继续以三段论为例,其基本结构包括大前提,小前提,结论三个部分,在这三个部分中大前提,小前提是已知的知识,而结论则是通过已知的知识所推理出来的新的知识。
随着信息数据爆炸式增长,以数据的分析、深度挖掘和融合应用为主要特征的数据时代已经来临。数据分析是有组织有目的地收集数据、分析数据,使之成为信息的过程。数据分析的目的是把隐藏在一大批看来杂乱无章的数据中的信息集中和提炼出来,从而找出所研究对象的内在规律。在实际应用中,数据分析可帮助做出判断,以便采取适当行动。
现有技术中,如公开号CN114822698A公开了一种基于知识推理的生物学大样本数据集分析方法及系统,该方法采用逻辑数学的方法分析多个变量之间的协同作用对结果产生的影响,有效地发现隐含在数据中的特定的关系,推测数据的发展趋势。该方法采用的技术方案为:收集m个案例的第一序列信息;定义条件变量和结果变量,数据集编码;计算单个条件变量的必要性指数;条件变量统计学检验;最小化推理及案例支持率计算;条件组合统计学检验;迭代计算;本生物学大样本数据集数据所得到的解为每次计算所得到的结果的集合;系统包括:包括序列检测模块,编码模块,必要性指数计算模块,条件变量统计学检验模块,最小化推理模块,条件组合案例支持率计算模块,条件组合统计学检验模块,迭代计算管理模块;装置包括:测序仪,存储器,处理器。
在计算了大量模拟数据、真实数据之后,发现该方法存在编码复杂,如果条件变量较多,则计算耗时很长,对计算机算力要求更高,迭代计算中删除的信息的量小造成迭代次数较多,得到的条件变量的组合不够简约的问题。
发明内容
为了解决现有技术中存在的问题,本发明提供了一种迭代分析生物学大样本数据的知识推理方法及系统,其采用二元编码,首先对条件变量进行筛选,然后再进行迭代计算,包括最小化推理,模型提取,统计学计算,模型覆盖的条件变量的编码的删除,形成新的用于迭代计算的数据集,分析多个变量组合成的模型对结果产生的影响,快速发现隐含的、有效的、有价值的、可理解的简约的适用性强的模型,得出待研究问题的趋向和关联。
为解决现有技术的问题,本发明提出了一种迭代分析生物学大样本数据的知识推理方法、系统及装置,包括:
一种迭代分析生物学大样本数据的知识推理方法,针对待分析的问题,收集m个案例的第一序列信息,所述第一序列信息采样于所述案例的同一位置的基因片段;
从所述基因片段中选取n个等位基因定义为条件变量,根据待分析的问题定义结果变量,将每个案例的所述条件变量和结果变量编码,将野生型纯合子基因型编码为0,将杂合子基因型以及突变纯合子基因型编码为1,将显性结果编码为1,将隐性结果编码为0,形成以数据矩阵表示的生物学大样本数据集,其每一行代表一个案例;
根据单个条件变量的状态Sx和结果变量的状态Sy计算必要性指数Nec:
Nec=Num(Sx,Sy)/Num(Sx),
其中,Num(Sx,Sy)表示条件变量的状态为Sx且结果变量的状态为Sy的案例的数量,Num(Sx) 表示条件变量的状态为Sx的案例的数量;
筛选出所有必要性指数Nec的值大于或等于第一预设值的条件变量Vxi,从数据矩阵表示的生物学大样本数据集中选取所有的样本及对应的结果变量Vy构建新的生物学大样本数据集,新的生物学大样本数据集包含经过筛选的所有条件变量的编码;
将新的生物学大样本数据集的数据复制到用于迭代计算的数据集中;
对用于迭代计算的数据集进行最小化推理得到不同的条件变量的组合Ci并计算其对应的组合解释力Exp,对组合Ci根据其组合解释力进行降序排列;
从第一个组合开始,提取编码为1的条件变量,将这些条件变量组合成模型Mi;如果组合Ci中所有条件变量均为0,则选取组合解释力Exp第二大的组合,依次类推;如果所有的条件变量的组合都提取不到模型,则整个迭代计算停止,将前面几轮迭代计算得到的结果输出;
在本轮用于迭代计算的数据集中进行模型Mi的统计学计算,统计学计算采用皮尔森卡方检验,将计算得到的P值添加至模型Mi之后,将模型Mi及其P值添加至结果集合中;
如果P值小于或等于0.05,则通过统计学检验;如果P值大于0.05,则未通过统计学检验;
在本轮用于迭代计算的数据集中删除模型Mi所覆盖的条件变量及每个样本所对应的编码,形成新的用于迭代计算的数据集;
将新的用于迭代数据集进行最小化推理,模型提取,统计学计算,模型覆盖的条件变量的编码的删除,形成新的用于迭代计算的数据集;
进行迭代计算,得到结果集合。
根据本发明的另一个方面,所述收集m个案例的第一序列信息步骤中,每个案例的第一序列信息收集的步骤包括:将每平方厘米点阵密度高于400的探针分子固定于支持物上后与标记的样品分子进行杂交,检测每个探针分子的杂交信号强度获取样品分子的序列信息。
根据本发明的另一个方面,所述最小化推理包括以下步骤:
S1:选出结果变量为状态Sy的组合项,删除重复的组合项,生成新表;
S2:在新表中,将单个组合项按含0个状态“0”,含1个状态“0”,含2个状态“0”,直至含n个状态“0”划分为不同的组,并按状态“0”的数量降序排列成表,其中n为条件变量个数;
S3: 准备一张新表,从含有最多数量的状态“0”的组开始依次向下,将当前组中的每一个组合项与下一组的每一个组合项比较,若两个组合项只有一个不同的条件变量,则将所述不同的条件变量用两个组合项所包含的两种不同的状态标记提取出来形成数列,所述数列代表一种新的状态,如果所述数列包含了所述不同的条件变量的所有取值或包含了“-”标记,则所述不同的条件变量用“-”标记,“-”标记代表对应的条件变量已消去,其可以取所有已编码的值,用状态相同的条件变量加上所述数列按照初始的条件变量的顺序生成一个新的组合项;如果新的组合项在新表中不存在,则将这个新的组合项放入新表中;如果新的组合项在新表中已存在,则不执行放入动作;
S4: 在新表中,重复步骤S2,S3直到新表中不存在只有一个条件变量不同的组合项为止。
根据本发明的另一个方面,得到组合解释力Exp的计算方式为:
Exp=Num(Sc,Sy)/Num(Sy),
其中,Num(Sc,Sy)表示条件变量组合为状态Sc且结果变量状态同时为Sy的案例的数量,Num(Sy) 表示结果变量为状态Sy的案例的数量。
一种迭代分析生物学大样本数据的知识推理系统,其特征在于:
包括序列检测模块,编码模块,必要性指数计算模块,新数据集生成模块,最小化推理模块,组合解释力计算模块,模型生成模块,模型统计学计算模块,迭代计算管理模块;
所述序列检测模块用于针对待分析的问题,收集m个案例的第一序列信息,所述第一序列信息采样于所述案例的同一位置的基因片段;
所述编码模块用于从所述基因片段中选取n个等位基因定义为条件变量,根据待分析的问题定义结果变量,将每个案例的所述条件变量和结果变量编码,将野生型纯合子基因型编码为0,将杂合子基因型以及突变纯合子基因型编码为1,将显性结果编码为1,将隐性结果编码为0,形成以数据矩阵表示的生物学大样本数据集,其每一行代表一个案例;
所述必要性指数计算模块用于计算单个条件变量的状态Sx相对于结果变量的状态Sy的必要性指数Nec,Nec=Num(Sx,Sy)/Num(Sx),其中,Num(Sx,Sy)表示条件变量的状态为Sx且结果变量的状态为Sy的案例的数量,Num(Sx) 表示条件变量的状态为Sx的案例的数量;
所述新数据集生成模块筛选出所有必要性指数Nec的值大于或等于第一预设值的条件变量Vxi,从数据矩阵表示的生物学大样本数据集中选取所有的样本及对应的结果变量Vy构建新的生物学大样本数据集,新的生物学大样本数据集包含经过筛选的所有条件变量的编码;
所述最小化推理模块用于对所述生物学大样本数据集进行最小化推理得到不同的条件变量的组合Ci;
所述组合解释力计算模块用于计算Ci的组合解释力Exp, Sup=Num(Sc,Sy)/Num(Sy),其中,Num(Sc,Sy)表示条件变量组合为状态Sc且结果变量状态同时为Sy的案例的数量,Num(Sy) 表示结果变量为状态Sy的案例的数量;
所述模型生成模块提取条件组合中编码为1的条件变量,将这些条件变量组合成模型Mi;
所述模型统计学计算模块采用皮尔森卡方检验,将计算得到的P值添加至模型Mi之后,将模型Mi及其P值添加至结果集合中;
所述迭代计算管理模块用于管理迭代计算,得到结果集合。
根据本发明的另一个方面,所述最小化推理包括以下步骤:
S1:选出结果变量为状态Sy的组合项,删除重复的组合项,生成新表;
S2:在新表中,将单个组合项按含0个状态“0”,含1个状态“0”,含2个状态“0”,直至含n个状态“0”划分为不同的组,并按状态“0”的数量降序排列成表,其中n为条件变量个数;
S3: 准备一张新表,从含有最多数量的状态“0”的组开始依次向下,将当前组中的每一个组合项与下一组的每一个组合项比较,若两个组合项只有一个不同的条件变量,则将所述不同的条件变量用两个组合项所包含的两种不同的状态标记提取出来形成数列,所述数列代表一种新的状态,如果所述数列包含了所述不同的条件变量的所有取值或包含了“-”标记,则所述不同的条件变量用“-”标记,“-”标记代表对应的条件变量已消去,其可以取所有已编码的值,用状态相同的条件变量加上所述数列按照初始的条件变量的顺序生成一个新的组合项;如果新的组合项在新表中不存在,则将这个新的组合项放入新表中;如果新的组合项在新表中已存在,则不执行放入动作;
S4: 在新表中,重复步骤S2,S3直到新表中不存在只有一个条件变量不同的组合项为止。
根据本发明的另一个方面,得到组合解释力Exp的计算方式为:
Exp=Num(Sc,Sy)/Num(Sy),
其中,Num(Sc,Sy)表示条件变量组合为状态Sc且结果变量状态同时为Sy的案例的数量,Num(Sy) 表示结果变量为状态Sy的案例的数量。
一种迭代分析生物学大样本数据的知识推理的装置,其特征在于,包括:
测序仪,用于基因测序;
存储器,用于存储程序;
处理器,用于加载程序,以执行如上文所述的迭代分析生物学大样本数据的知识推理方法。
本发明相比于现有技术具有如下有益效果:
本申请的发明点在于1、简化了编码,与现有技术中的“将每个案例的条件变量和结果变量以代表不同状态的阿拉伯数字以升序的方式进行编码,所述条件变量的状态不大于三种,所述结果变量的状态不大于两种”相比,本申请简化了编码方式,将野生型纯合子基因型编码为0,将杂合子基因型以及突变纯合子基因型编码为1,将显性结果编码为1,将隐性结果编码为0。该编码方式使得条件变量的取值仅限于0和1,在进行最小化推理时,更加有利于消去与结果无关的条件变量,使得到的条件变量的组合更加简约,提高了运算速度。
2采用并优化了筛选的方法,与现有技术中直接采用源数据进行计算相比,本申请首先先根据单个条件变量的状态Sx和结果变量的状态Sy计算必要性指数Nec,根据必要性指数筛选出所有必要性指数Nec的值大于或等于第一预设值的条件变量Vxi,从数据矩阵表示的生物学大样本数据集中选取所有的样本及对应的结果变量Vy构建新的生物学大样本数据集,新的生物学大样本数据集包含经过筛选的所有条件变量的编码,再用新的生物学大样本数据集进行迭代计算,包括最小化推理,模型提取,统计学计算,模型覆盖的条件变量的编码的删除,形成新的用于迭代计算的数据集);该筛选步骤可以快速地过滤掉结果变量的非必要条件,有效提高了运算速度。
3、采用并优化了变量迭代排除的方法,与现有技术中的“删除所述上一轮计算中添加至结果集合的组合所覆盖的案例形成新数据集”相比,变量迭代排除方法的迭代次数更少,迭代计算中删除的信息的量更大且更加合理,可以以更少的运算时间得到更加合理的结果。
4、采用并优化了模型提取的方法,从条件变量的组合中提取编码为1的条件变量,将这些条件变量组合成模型,与现有技术得到的条件变量的组合相比,结果更加简约,适用性更强。
5、通过本生物学大样本数据集数据所得到的解为每次计算提取到的模型的集合。该集合可以作为性状研究的基础材料数据,也可作为生物学网络的推理及构建的参考。在上述发明点的基础上得到本申请的技术方案。本发明采用以上技术方案与现有技术相比的有益效果是:简化编码使得到的条件变量的组合更加简约,提高了运算速度;可以快速地过滤掉结果变量的非必要条件,有效提高了运算速度;迭代计算中删除的信息的量更大且更加合理,可以以更少的运算时间得到更加合理的结果;在条件变量的组合的基础上进行模型提取,使得结果更加简约,适用性更强。能够迅速、高效地计算出生物学大样本数据集中反映出来的导致结果发生的多个条件变量的更加简约、适应性强的模型,有效地发现隐含在数据中的某些特定的关系,推测数据的发展趋势,作为性状研究的基础材料数据,也可作为生物学网络的推理及构建的参考。
附图说明
构成本发明创造的一部分的附图用来提供对本发明创造的进一步理解,本发明创造的示意性实施例及其说明用于解释本发明创造,并不构成对本发明创造的不当限定。在附图中:
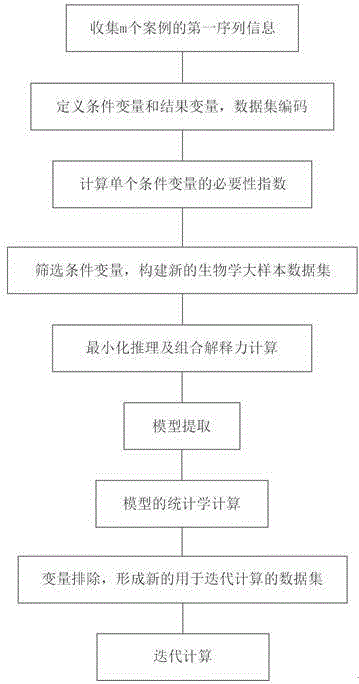
图1为本发明提供的一种迭代分析生物学大样本数据的知识推理方法的流程图。
图2为本发明提供的一种迭代分析生物学大样本数据的知识推理系统的结构图。
具体实施方式
下面将结合实施例对本发明的技术方案进行清楚、完整地描述。
除非特别说明,否则,本发明中的“第一”、“第二”等描述均用来区分不同的对象,并不用来表示大小或时序等含义,且不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”等的特征可以明示或者隐含地包括一个或者更多个该特征。在本发明创造的描述中,除非另有说明,“多个”的含义是两个或两个以上。
本发明中的术语“和/或”,仅是一种描述关联对象的关联关系,表示可以存在三种关系,例如,A和/或B,可以表示:单独存在A,单独存在B,和同时存在A和B这三种情况。显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
根据本发明的示范性实施例,图1图示一种迭代分析生物学大样本数据的知识推理方法的流程图。该种迭代分析生物学大样本数据的知识推理方法包括:
步骤一:收集m个案例的第一序列信息:针对待分析的问题,收集m个案例的第一序列信息,优选地,m取值为100或100以上;所述第一序列信息采样于所述案例的同一位置的基因片段;
步骤二:定义条件变量和结果变量,数据集编码:从所述基因片段中选取n个等位基因定义为条件变量,优选地,n取值为m除以10得到的结果的整数部分;从所述基因片段中选取n个等位基因定义为条件变量,根据待分析的问题定义结果变量,将每个案例的所述条件变量和结果变量编码,将野生型纯合子基因型编码为0,将杂合子基因型以及突变纯合子基因型编码为1,将显性结果编码为1,将隐性结果编码为0,形成以数据矩阵表示的生物学大样本数据集,其每一行代表一个案例;
步骤三:根据单个条件变量的状态Sx和结果变量的状态Sy计算必要性指数Nec:
Nec=Num(Sx,Sy)/Num(Sx),
其中,Num(Sx,Sy)表示条件变量的状态为Sx且结果变量的状态为Sy的案例的数量,Num(Sx) 表示条件变量的状态为Sx的案例的数量;
步骤四:筛选条件变量,构建新的生物学大样本数据集:筛选出所有必要性指数Nec的值大于或等于第一预设值的条件变量Vxi,从数据矩阵表示的生物学大样本数据集中选取所有的样本及对应的结果变量Vy构建新的生物学大样本数据集,新的生物学大样本数据集包含经过筛选的所有条件变量的编码;将新的生物学大样本数据集的数据复制到用于迭代计算的数据集中;
步骤五:最小化推理及组合解释力计算:对用于迭代计算的数据集进行最小化推理得到不同的条件变量的组合Ci并计算其对应的组合解释力Exp,对组合Ci根据其组合解释力进行降序排列;
步骤六:模型提取:从第一个组合开始,提取编码为1的条件变量,将这些条件变量组合成模型Mi;如果组合Ci中所有条件变量均为0,则选取组合解释力Exp第二大的组合,依次类推;如果所有的条件变量的组合都提取不到模型,则整个迭代计算停止,将前面几轮迭代计算得到的结果输出;
步骤七:模型的统计学计算:在本轮用于迭代计算的数据集中进行模型Mi的统计学计算,统计学计算采用皮尔森卡方检验,将计算得到的P值添加至模型Mi之后,将模型Mi及其P值添加至结果集合中;
如果P值小于或等于0.05,则通过统计学检验;如果P值大于0.05,则未通过统计学检验;
步骤八:变量排除,形成新的用于迭代计算的数据集:在本轮用于迭代计算的数据集中删除模型Mi所覆盖的条件变量及每个样本所对应的编码,形成新的用于迭代计算的数据集;
将新的用于迭代数据集进行最小化推理,模型提取,统计学计算,模型覆盖的条件变量的编码的删除,形成新的用于迭代计算的数据集;
进行迭代计算,得到结果集合。
所述收集m个案例的第一序列信息步骤中,每个案例的第一序列信息收集的步骤包括:将每平方厘米点阵密度高于400的探针分子固定于支持物上后与标记的样品分子进行杂交,检测每个探针分子的杂交信号强度获取样品分子的序列信息。
所述最小化推理包括以下步骤:
S1:选出结果变量为状态Sy的组合项,删除重复的组合项,生成新表;
S2:在新表中,将单个组合项按含0个状态“0”,含1个状态“0”,含2个状态“0”,直至含n个状态“0”划分为不同的组,并按状态“0”的数量降序排列成表,其中n为条件变量个数;
S3: 准备一张新表,从含有最多数量的状态“0”的组开始依次向下,将当前组中的每一个组合项与下一组的每一个组合项比较,若两个组合项只有一个不同的条件变量,则将所述不同的条件变量用两个组合项所包含的两种不同的状态标记提取出来形成数列,所述数列代表一种新的状态,如果所述数列包含了所述不同的条件变量的所有取值或包含了“-”标记,则所述不同的条件变量用“-”标记,“-”标记代表对应的条件变量已消去,其可以取所有已编码的值,用状态相同的条件变量加上所述数列按照初始的条件变量的顺序生成一个新的组合项;如果新的组合项在新表中不存在,则将这个新的组合项放入新表中;如果新的组合项在新表中已存在,则不执行放入动作;
S4: 在新表中,重复步骤S2,S3直到新表中不存在只有一个条件变量不同的组合项为止。
得到组合解释力Exp的计算方式为:Exp=Num(Sc,Sy)/Num(Sc),其中,Num(Sc,Sy)表示条件变量组合为状态Sc且结果变量状态同时为Sy的案例的数量,Num(Sc) 表示条件变量组合为状态Sc的案例的数量。
根据本发明的示范性实施例,图2图示一种迭代分析生物学大样本数据的知识推理系统的结构图。该种迭代分析生物学大样本数据的知识推理系统包括:序列检测模块,编码模块,必要性指数计算模块,新数据集生成模块,最小化推理模块,组合解释力计算模块,模型生成模块,模型统计学计算模块,迭代计算管理模块;
所述序列检测模块用于针对待分析的问题,收集m个案例的第一序列信息,所述第一序列信息采样于所述案例的同一位置的基因片段;
所述编码模块用于从所述基因片段中选取n个等位基因定义为条件变量,根据待分析的问题定义结果变量,将每个案例的所述条件变量和结果变量编码,将野生型纯合子基因型编码为0,将杂合子基因型以及突变纯合子基因型编码为1,将显性结果编码为1,将隐性结果编码为0,形成以数据矩阵表示的生物学大样本数据集,其每一行代表一个案例;
所述必要性指数计算模块用于计算单个条件变量的状态Sx相对于结果变量的状态Sy的必要性指数Nec,Nec=Num(Sx,Sy)/Num(Sx),其中,Num(Sx,Sy)表示条件变量的状态为Sx且结果变量的状态为Sy的案例的数量,Num(Sx) 表示条件变量的状态为Sx的案例的数量;
所述新数据集生成模块筛选出所有必要性指数Nec的值大于或等于第一预设值的条件变量Vxi,从数据矩阵表示的生物学大样本数据集中选取所有的样本及对应的结果变量Vy构建新的生物学大样本数据集,新的生物学大样本数据集包含经过筛选的所有条件变量的编码;
所述最小化推理模块用于对所述生物学大样本数据集进行最小化推理得到不同的条件变量的组合Ci;
所述组合解释力计算模块用于计算Ci的组合解释力Exp, Sup=Num(Sc,Sy)/Num(Sy),其中,Num(Sc,Sy)表示条件变量组合为状态Sc且结果变量状态同时为Sy的案例的数量,Num(Sy) 表示结果变量为状态Sy的案例的数量;
所述模型生成模块提取条件组合中编码为1的条件变量,将这些条件变量组合成模型Mi;
所述模型统计学计算模块采用皮尔森卡方检验,将计算得到的P值添加至模型Mi之后,将模型Mi及其P值添加至结果集合中;
所述迭代计算管理模块用于管理迭代计算,得到结果集合。
所述最小化推理包括以下步骤:
S1:选出结果变量为状态Sy的组合项,删除重复的组合项,生成新表;
S2:在新表中,将单个组合项按含0个状态“0”,含1个状态“0”,含2个状态“0”,直至含n个状态“0”划分为不同的组,并按状态“0”的数量降序排列成表,其中n为条件变量个数;
S3: 准备一张新表,从含有最多数量的状态“0”的组开始依次向下,将当前组中的每一个组合项与下一组的每一个组合项比较,若两个组合项只有一个不同的条件变量,则将所述不同的条件变量用两个组合项所包含的两种不同的状态标记提取出来形成数列,所述数列代表一种新的状态,如果所述数列包含了所述不同的条件变量的所有取值或包含了“-”标记,则所述不同的条件变量用“-”标记,“-”标记代表对应的条件变量已消去,其可以取所有已编码的值,用状态相同的条件变量加上所述数列按照初始的条件变量的顺序生成一个新的组合项;如果新的组合项在新表中不存在,则将这个新的组合项放入新表中;如果新的组合项在新表中已存在,则不执行放入动作;
S4: 在新表中,重复步骤S2,S3直到新表中不存在只有一个条件变量不同的组合项为止。
得到组合解释力Exp的计算方式为:
Exp=Num(Sc,Sy)/Num(Sy),
其中,Num(Sc,Sy)表示条件变量组合为状态Sc且结果变量状态同时为Sy的案例的数量,Num(Sy) 表示结果变量为状态Sy的案例的数量。
根据本发明的示范性实施例,本发明还提供了一种迭代分析生物学大样本数据的知识推理的装置,其特征在于,包括:
测序仪,用于基因测序;
存储器,用于存储程序;
处理器,用于加载程序,以执行如上文所述的迭代分析生物学大样本数据的知识推理方法。
将本申请中的方法简称为方法一,将申请号为CN114822698A中公开的一种基于知识推理的生物学大样本数据集分析方法及系统的分析方法简称为方法二,通过将两种方法进行编程,分析同一数据集,比较得到的结果、已公开文献对结果的支持率及运算时长。第i个条件变量记为CV
实施例1
数据集1包含121个案例,其中结果变量为显性的63例,结果变量为隐性的58例,条件变量个数为13。经过计算,比较结果见表1。
表1 数据集1的分析结果比较
方法一与方法二均计算得到了4个组合,均得到已公开文献的支持,方法一运算时长仅为方法二的15.5%。方法一的运算速度更快,效率更高。
实施例2
数据集2包含336个案例,其中结果变量为显性的223例,结果变量为隐性的113例,条件变量个数为56。经过计算,比较结果见表2。
表2 数据集2的分析结果比较
方法一获得了4项结果,已公开文献支持率为75%,方法二获得了7项结果,已公开文献支持率为71.4%,方法一运算时长仅为方法二的15%,说明方法一的能得到更多有公开文献的支持的结果,运算速度更快,效率更高。
实施例3
数据集3包含593个案例,其中结果变量为显性的431例,结果变量为隐性的162例,条件变量个数为106。经过计算,比较结果见表3。
表3 数据集3的分析结果比较
方法一获得了7项结果,已公开文献支持率为85.7%,方法二获得了10项结果,已公开文献支持率为70%,方法一运算时长仅为方法二的17.6%,说明方法一的准确率更高,运算速度更快,效率更高。
实施例4
数据集4包含861个案例,其中结果变量为显性的592例,结果变量为隐性的269例,条件变量个数为205。经过计算,比较结果见表4。
表4 数据集4的分析结果比较
方法一获得了9项结果,已公开文献支持率为88.9%,方法二获得了11项结果,已公开文献支持率为72.7%,方法一运算时长仅为方法二的17.4%,说明方法一的准确率更高,运算速度更快,效率更高。
实施例5
数据集5包含962个案例,其中结果变量为显性的631例,结果变量为隐性的331例,条件变量个数为262。经过计算,比较结果见表5。
表5 数据集5的分析结果比较
方法一获得了10项结果,已公开文献支持率为90%,方法二获得了10项结果,已公开文献支持率为80%,方法一运算时长仅为方法二的19.6%,说明方法一的准确率更高,运算速度更快,效率更高。
实施例6
数据集6包含1073个案例,其中结果变量为显性的832例,结果变量为隐性的241例,条件变量个数为281。经过计算,比较结果见表6。
表6 数据集6的分析结果比较
方法一获得了12项结果,已公开文献支持率为91.7%,方法二获得了14项结果,已公开文献支持率为85.7%,方法一运算时长仅为方法二的16.5%,说明方法一的准确率更高,运算速度更快,效率更高。
应当注意的是:因为本发明解决了现有分析方法的运算速度较慢,分析结果不简约,适用性不强的问题,通过简化编码使得到的条件变量的组合更加简约,提高了运算速度;可以快速地过滤掉结果变量的非必要条件,有效提高了运算速度;迭代计算中删除的信息的量更大且更加合理,可以以更少的运算时间得到更加合理的结果;在条件变量的组合的基础上进行模型提取,使得结果更加简约,适用性更强。能够迅速、高效地计算出生物学大样本数据集中反映出来的导致结果发生的多个条件变量的模型,有效地发现隐含在数据中的某些特定的关系。采用了计算机技术领域中技术人员在阅读本说明书之后根据其教导所能理解的技术手段,并获得迅速、高效地计算出生物学大样本数据集中反映出来的导致结果发生的多个条件变量的更加简约、适应性强的模型,有效地发现隐含在数据中的某些特定的关系,推测数据的发展趋势的有益技术效果,所以在所附权利要求中要求保护的方案属于专利法意义上的技术方案。
以上所述,仅为本发明的较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到的变化或替换,都应包涵在本发明的保护范围之内。除非以其他方式明确陈述,否则公开的每个特征仅是一般系列的等效或类似特征的一个示例。因此,本发明的保护范围应该以权利要求书的保护范围为准。