含能分子设计的高通量计算方法与系统
文献发布时间:2024-04-18 19:57:50
技术领域
本发明涉及化学信息学中的含能分子设计技术领域,特别是一种含能分子设计的高通量计算方法与系统。
背景技术
含能分子是军用炸药、发射药和火箭推进剂配方的重要组成部分,在国防、航天、民用都有广泛的应用。含能分子必须满足能量、感度、热稳定性、吸湿、水解、力学强度等多方面性能需求,导致其研制链条长、环节多。受此影响,含能分子的更新换代极具挑战性。随着计算机辅助分子设计在含能分子的研究中逐步获得应用,以计算机模型提前预测重要的性能,可以为合成化学家推荐有前景的目标分子,使含能分子的研制效率在一定程度上得以提升。随着含能分子设计研究的不断深入,计算方法和程序存在的不足也逐步显现出来,其便捷性、专用性、自动化程度尚无法满足大数据量的计算和成批量分子筛选的需求。
近年来,伴随材料基因组计划的推进,高通量计算、大数据和人工智能等新技术在金属和无机材料设计方面已获得诸多可喜的成绩。AFLOW、Material Project、MatCloud等一些高通量计算软件平台陆续被报道,它们通过自动化的流程和高效并发的计算实现了大批量化学结构的建模和性能计算,能够在相对较短时间内从数百万无标记结构中发现新的目标化合物。高通量计算将繁琐的程序与方法集成到易于使用的系统中,从而使得不具备跨学科知识的普通研究人员都能轻松完成材料设计。由于高通量计算可处理的任务量较传统的“画加算”模式高出好几个数量级,可产生“海量”的化学结构和性能数据,与数据挖掘算法结合后可形成数据驱动型的材料设计新模式。
当前,高通量计算技术在含能分子设计方面的应用面临困难,阻碍了数据驱动型的含能分子设计的开展。其根本的原因在于含能分子应用性能的预测在原理上不同于无机材料和金属材料。很多材料的应用性能可以通过量子化学计算的结果进行直接的关联,比如用带隙评估光伏电池的效率或以吸附能大小筛选合金催化剂。预测含能分子的应用性能,除了需要量子化学计算,还要再经过多步构效关系模型的递推才能实现。含能分子的应用性能是从分子拓扑出发到工程应用特性评价的多学科交叉问题,涉及多种门类的方法、程序和复杂的应用流程。这需要通过大量人机交互实现多个方法和程序的串联,只有具备多学科知识和丰富分子设计经验的专业人员才能开展相关工作,技术门槛很高。因此,面向含能分子设计开发高通量设计流程与系统具有重要意义,可以显著降低新方法新技术应用的知识门槛,以数据驱动型的设计加速发现新型含能分子。
发明内容
为解决现有技术中存在的问题,本发明的目的是提供一种含能分子设计的高通量计算方法与系统,本发明实现了高效、快速的从头设计并筛选含能分子的目的。
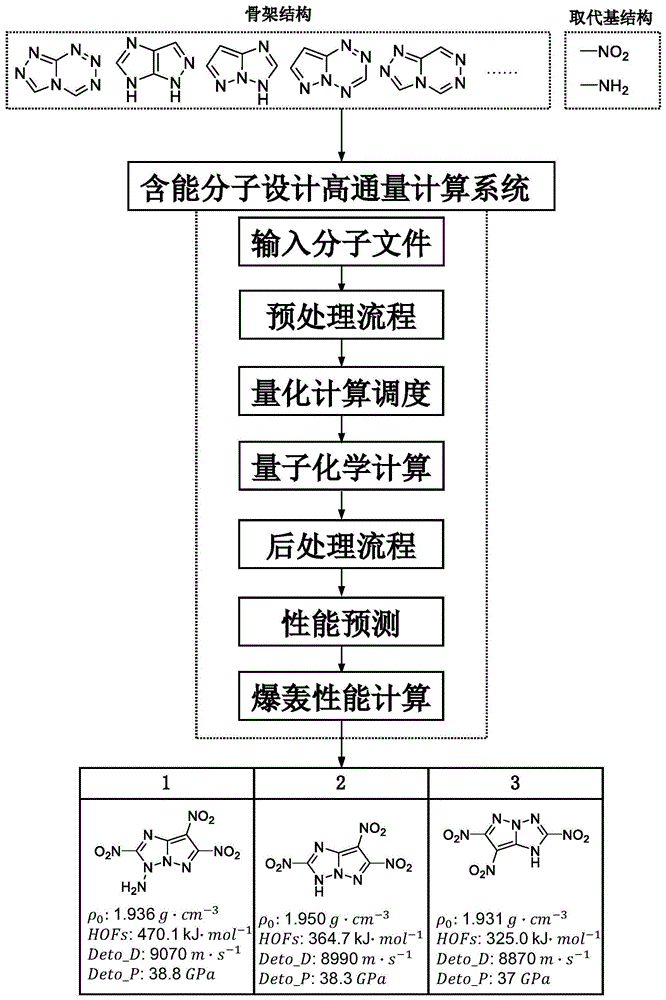
为实现上述目的,本发明采用的技术方案是:一种含能分子设计的高通量计算方法,包括:
输入分子文件:将输入的骨架及取代基结构排列组合生成可能的结构文件,并去除重复的结构文件;
预处理流程:将生成的大量输入分子文件根据用户计算需求,构象搜索并生成高斯计算文件;
量化计算调度:启动多队列调度量子化学计算软件开展多任务并行计算,随机抓取高斯计算文件进行量子化学计算,高斯计算文件全部计算完成后自动结束;
后处理流程:获取并分析量子化学计算结果,得到化合物的本征属性,并将其作为性能预测的描述符;
性能预测:根据以含能分子的本征属性为分子描述符构建的性能预测模型完成含能分子的物理化学属性预测,得到化合物的物理化学属性并作为爆轰性能计算的输入信息;
爆轰性能计算:根据爆轰状态方程进行爆轰方程计算,得到化合物的应用属性。
作为本发明的进一步改进,在输入分子文件时,用户通过绘制骨架结构并以smiles字符串给定取代基结构,输入分子文件对骨架及取代基结构进行排列组合并去除重复结构得到所有可能的结构文件,并以mol文件格式给出。
作为本发明的进一步改进,所述预处理流程具体包括导入分子文件、任务设置、参数设置及生成高斯计算文件,具体如下:
用户导入计算的分子文件并根据计算需求选择合适的计算水平、内存大小、是否需要计算键解离能的输入任务信息,根据用户导入的分子文件完成构象搜索并基于能量最有利的构象自动生成相应的高斯计算文件;其中,构象搜索包括构象采样、几何优化和能量排序。
作为本发明的进一步改进,在量化计算调度时,用户自定义启动m个队列,每个队列每次随机抓取一个计算文件并调度Gauss ian软件开始量化计算,包括结构优化、频率计算以及单点能量计算;每个队列中的文件计算完成后自动提取下一个新的文件,直至计算文件全部完成。
作为本发明的进一步改进,在所述后处理流程中,静电势分析采用Multiwfn读取量子化学计算结果得到含能分子的本征属性,并且作为分子描述符用于作为性能预测环节的输入信息用于构建性能预测模型;得到的含能分子的本征属性包括:分子表面积,偶极矩,电荷分离度,球度,分子体积,分子平面性指数,分子量,原子体积,分子形状,标准状态下的热力学焓值。
作为本发明的进一步改进,所述的性能预测流程中,所述物理化学属性包括:气相生成焓、升华焓、键解离能、固相生成焓、晶体密度、及元素组成;其中气相生成焓基于CBS-4M计算水平得到的298K下的热焓值H
其中ρ为密度,ΔH
作为本发明的进一步改进,所述爆轰状态方程包括Kamei-Jacob方程、Jones-Wilkins-Lee方程、Vortex Lattice Method方程和Beckker-Kistiakowsk-Wi lson方程,用户选择合适的爆轰状态方程用以计算含能分子爆轰性能,即应用属性;可供得到的应用属性包括:爆热,爆压(P),爆速(D),爆炸体积,爆炸温度。
作为本发明的进一步改进,将所述本征属性、物理化学属性、应用属性以Excel表格的方式保存,便于用户查看并筛选判断。
本发明还提供一种含能分子设计的高通量计算系统,包括:
输入分子文件模块:用于将输入的骨架及取代基结构排列组合生成可能的结构文件,并去除重复的结构文件;
预处理流程模块:用于将生成的大量输入分子文件根据用户计算需求,构象搜索并生成高斯计算文件;
量化计算调度模块:用于启动多队列调度量子化学计算软件开展多任务并行计算,随机抓取高斯计算文件进行量子化学计算,高斯计算文件全部计算完成后自动结束;
后处理流程模块:用于获取并分析量子化学计算结果,得到化合物的本征属性,并将其作为性能预测的描述符;
性能预测模块:用于根据以含能分子的本征属性为分子描述符构建的性能预测模型完成含能分子的物理化学属性预测,得到化合物的物理化学属性并作为爆轰性能计算的输入信息;
爆轰性能计算模块:用于根据爆轰状态方程进行爆轰方程计算,得到化合物的应用属性。
本发明的有益效果是:
本发明采用面向数据驱动型含能分子设计的高通量计算流程,兼顾了数据分析、量化计算、数据处理、性能预测、爆轰计算流程,是一套给用户提供了高效、快速的从头设计并筛选含能分子的自动化系统。用户给定骨架及取代基通过输入分子文件生成大量可能的分子结构,通过预处理流程生成对应的计算文件而后进行量子化学计算,后处理流程分析量子化学计算结果得到化合物的本征属性,进一步通过性能预测环节得到化合物的物理化学属性和通过爆轰性能计算得到化合物的应用属性。该方法全过程智能化系统化处理数据,无需要求用户具备多学科的专业知识,降低了应用门槛,有利于分子设计普及化。除此之外,用户还可以借助性能预测板块及爆轰性能计算预测已有的含能分子的各项性能,可为实验研发提供参考价值。
附图说明
图1为本发明实施例2完成含氮五元并环含能分子高通量设计流程图;
图2为本发明实施例3完成稠环类含能分子高通量设计流程图;
图3为本发明实施例4完成氮杂稠环含能分子高通量设计流程图;
图4为本发明实施例5完成二联唑类含能分子高通量设计流程图。
具体实施方式
下面结合附图对本发明的实施例进行详细说明。
实施例1
本实施例提供面向数据驱动型含能分子设计的高通量计算方法,本实施例本着从头设计的理念,基于量子化学计算得到含能分子的本征属性,并进一步通过原子化方案、构效关系模型分析得到物理化学属性,从而选择合适的爆轰状态方程计算得到应用性能完成高通量计算流程。综合考虑三个方面的属性完成对含能分子的性能预测,提出面向含能分子的高通量计算系统,从而实现对含能分子的高通量设计,为新含能化合物的设计提供一些理论依据。
利用计算机自动生成大量的计算文件,基于量子化学计算及性能预测模型对所生成的含能分子结构的应用属性进行预测,完成新型含能分子设计的高通量计算流程,从而加速新型含能分子的研发,该方法包括:
输入分子文件,用户输入的骨架及取代基结构排列组合生成可能的结构文件,去除重复的结构;
预处理流程,用于处理生成的大量输入的分子文件根据用户计算需求,构象搜索并生成高斯计算文件;
量化计算调度,用于启动多队列调度量子化学计算软件开展多任务并行计算,随机抓取计算文件进行量子化学计算,计算文件全部计算完成后自动结束。
后处理流程,用于获取并分析量子化学计算结果,所得到的信息为化合物的本征属性并作为性能预测板块的分子描述符;
性能预测,用于根据以含能分子的本征属性为分子描述符构建的性能预测模型完成含能分子的物理化学属性计算,所得到的信息为化合物的物理化学属性并作为爆轰性能预测板块的输入信息;
爆轰性能计算,用于根据爆轰状态方程进行爆轰方程计算,所得到的信息为化合物的应用属性。
所述的输入分子文件,用户通过绘制骨架结构并以smi les字符串给定取代基结构,输入分子文件板块会对骨架及取代基结构进行排列组合并去除重复结构得到所有可能的结构文件,以mol文件格式给出。
所述的预处理流程,包括导入分子文件、任务设置、参数设置及生成计算文件等功能。用户导入计算的分子文件并根据计算需求选择合适的计算水平、内存大小、是否需要计算键解离能等输入任务信息,预处理流程会根据用户导入的分子文件完成构象搜索并基于能量最有利的构象自动生成与输入分子对应的高斯计算文件。其中,构象搜索包括构象采样、几何优化和能量排序。
所述的量化计算调度,用户自定义启动m个队列,每个队列每次随机抓取一个计算文件并调度Gauss ian软件开始量化计算,包括结构优化、频率计算以及单点能量计算。每个队列中的文件计算完成后自动提取下一个新的文件,直至计算文件全部完成。
所述的后处理流程,系统中静电势分析采用Mul tiwfn读取量子化学计算结果得到含能分子的本征属性,并且作为分子描述符用于作为性能预测环节的输入信息用于构建性能预测模型。
所述的含能分子的本征属性包括但不限于:分子表面积,偶极矩,电荷分离度,球度,分子体积,分子平面性指数,分子量,原子体积,分子形状,标准状态下的热力学焓值。
所述的性能预测环节,系统根据提取到的化合物的本征属性作为分子描述符通过构效关系模型得到含能分子的物理化学属性。
所述的含能分子的物理化学属性包括但不限于:气相生成焓、升华焓、键解离能、固相生成焓、晶体密度、及元素组成。其中气相生成焓基于CBS-4M计算水平得到的298K下的热焓值H
其中ρ为密度,ΔH
所述的爆轰性能计算环节,系统提供四个可供选择的爆轰状态方程,分别是Kamei-Jacob方程(简称K-J方程),Jones-Wi lkins-Lee方程(简称JWL方程),VortexLattice Method(简称VLM方程),Beckker-Kistiakowsk-Wi lson方程(简称BKW方程),用户可选择合适的方程用以计算含能分子爆轰性能,即应用属性。例如,K-J方程计算过程如下:
其中N为每克炸药爆轰生成气体的摩尔数,mol·g
所述的应用属性包括但不限于:爆热,爆压(P),爆速(D),爆炸体积,爆炸温度。
上述所有环节得到的化合物属性值均会以Excel表格的方式保存下来,便于用户查看并筛选判断。
实施例2
一般来说,含能分子的筛选过程需考虑包括但不限于键解离能、密度、生成焓、爆速、爆压等属性值,他们分别属于本征属性、物理化学属性以及应用属性三个方面的性质。如图1所示,以含氮五元并环为骨架,以硝基(-NO
通过本实施例可以并行处理数据驱动设计产生的大量分子文件且全过程自动化调度计算软件处理,尽管各个操作环节及计算原理需要开发者具备较高的专业知识高,理论基础扎实,但仅需用户提供计算所需的文件并指定计算需求,流程简单易操作,结果清晰明了,降低了新技术新方法的应用门槛,可以广泛推广使用。
实施例3
如图2所示,以稠环类含能化合物为研究对象,通过识别大量的高密度含能分子进行骨架预筛选获得高密度的稠环分子骨架,以-NO
实例证明基于高密度骨架开展高密度含能分子设计是一种可实施方案,并且通过输入分子文件环节可以得到7000多个含能分子,有利于开展含能分子设计。预处理流程与后处理流程为用户开展量子化学计算提供了快速、简单的操作模式,极大地提高了工作效率,提高了软件的泛化性。性能预测结合了化学计量学与量子化学计算保证了性能预测的准确性,为含能分子设计提供了理论基础。爆轰性能板块提供4个可供选择的爆轰状态方程,充分利用前两部分属性为含能分子筛选提供多层次参考指标,保证了面向数据驱动型含能分子设计高通量计算流程与系统的可行性。
实施例4
如图3所示,以三种氮杂稠环分子和两种联唑分子为基本骨架,以-NO
实施例5
如图4所示,以1H-1,2,4-三唑和1H-四唑构筑的二联唑为分子骨架,以-NH
以上所述实施例仅表达了本发明的具体实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
- 一种基于机器学习性能预测含能分子计算机辅助设计系统
- 一种基于机器学习性能预测含能分子计算机辅助设计系统