一种基于单细胞分析建立的用于评估头颈鳞癌预后的评分系统
文献发布时间:2024-04-18 19:58:21
技术领域
本发明涉及生物信息技术领域,具体涉及一种基于单细胞分析建立的用于评估头颈鳞癌预后的评分系统。
背景技术
头颈部鳞状细胞癌(head and neck squamous cell carcinoma,HNSCC)是头颈部最常见恶性肿瘤,也是全身第六大恶性肿瘤,每年新发病例约90万例,死亡病例约50万例。尽管有手术、放疗、化疗和免疫治疗等多种干预方式,但预后仍较差。进一步探索HNSCC发展机制,鉴定新的分子生物标志物作为更高效治疗靶点,对改善HNSCC患者预后具有重要意义。
能量代谢的重编程已被认为是肿瘤的普遍现象,是肿瘤细胞维持快速增殖的必要手段。恶性肿瘤细胞的代谢重编程是复杂且多步骤的过程,与许多分子改变有关。近年来,关于头颈鳞癌发生发展过程中代谢重编程的研究是领域内研究热点,糖酵解与氨基酸代谢等代谢途径的改变已被证实在HNSCC中发挥重要作用。
肿瘤组学分型是指通过基因组、转录组、蛋白质组、代谢组等组学技术,对肿瘤进行分子水平系统描绘,从多种层面探索肿瘤发生发展机制和内在特性,根据肿瘤分子特征表达谱进行肿瘤分型,并在此基础上对肿瘤进行更细致亚型分类,有利于改善肿瘤的诊断、预后分层、治疗指导、复发监控及药物研发。已有研究者对胰腺癌、宫颈癌、小儿脑癌、乳腺癌等进行了多种组学分子分型。
单细胞测序分析是指在单一细胞水平上进行的组学研究。近年来,作为前沿技术,单细胞测序分析广泛应用于肿瘤细胞异质性、肿瘤细胞转移机制、肿瘤干细胞等的研究,在理解肿瘤的发生、发展、转移以及耐药性机制方面起到了重要作用,为肿瘤的诊断和治疗提供了许多新的分子靶点和策略,应用该技术的研究高频次发表于高水平期刊上。
现有的HNSCC相关的单细胞测序分析构建的评分系统,由于来自scRNA-seq数据集的患者数量有限,很少有研究全面探讨细胞组成中反映的代谢重编程与患者异质性的联系。往往是根据某种特定的细胞比例,没有结合患者的预后情况,使其在临床上的应用受到限制。现有的HNSCC代谢组学构建的评分系统往往是基于某种特定的基因表达情况,或者是一群基因表达的建模,没有从细胞层面上描述代谢重编程对患者预后的影响,以及代谢重编程相关的评分系统。
发明内容
本发明通过对单细胞数据进行分析,获得包括高代谢活性细胞和低代谢活性细胞和整合的非恶性细胞组成,再利用反卷积算法,通过转录组数据上的基因信息来推算486名HNSCC患者的细胞组成结构。其中,高代谢活性细胞为具有高频细胞代谢重编程在scRNA数据中主要分布在转移样本的恶性细胞,将低代谢活性细胞则为具有相对较低的代谢活性、在scRNA数据中主要分布在原发样本的恶性细胞。定义高代谢活性细胞数据占所有细胞的比例为评分体系,由此构建出一种基于单细胞分析的可用于评估HNSCC预后的评分系统。
其中,基于单细胞分析建立的用于评估头颈鳞癌预后的评分方法包括:
获取样本数据,样本数据包括头颈鳞癌单细胞数据和大队列RNA数据,大队列RNA数据包括头颈鳞癌患者的RNA序列数据和临床信息;
对所述单细胞数据和大队列RNA数据进行质量控制;
对所述单细胞数据中的细胞进行聚类和注释;
定义恶性细胞和代谢活性基因;
根据代谢活性将细胞分群;
根据分群后的细胞代谢活性与头颈鳞癌的相关性,建立不同细胞群比例与头颈鳞癌预后的关系;
根据评分体系筛选代谢生物标志物。
进一步,对所述单细胞数据进行质量控制具体包括:
去除表达基因≤200的细胞及基因表达数≥2500的细胞;
去除双细胞;
去除表达在少于3个细胞中的基因;
去除线粒体基因;
合并数据集。
对大队列RNA数据进行质量控制可以包括:(1)保留未被福尔马林浸泡的样本;(2)保留有完整临床数据的样本;(3)保留有明确临床结局及生存时间的样本。
进一步,对所述单细胞数据中的细胞进行聚类和注释具体包括:
对所有细胞进行降维聚类,过滤掉每个亚群中核糖体基因表达超过35%的细胞;
寻找每一亚群中的高表达基因,并根据寻找出的高表达基因,特殊表达的基因和已报道过细胞标记基因对每一细胞亚群进行注释;
注释后的细胞分为上皮细胞、T细胞、B细胞、浆细胞、单核细胞、肥大细胞、成纤维细胞及恶性细胞等亚群。
进一步,所述定义恶性细胞包括:
取样本数据中的恶性细胞进行降维聚类;
根据每一群中恶性细胞的来源进行分类:
转移癌样本来源最多的细胞群定义为转移群,原发癌样本来源最多的细胞群定义为原发群,其余从转移癌和原发癌样本来源相当的恶性细胞群定义为混合群。
进一步,所述根据代谢活性将细胞分群包括:
计算代谢活性的截断值;
将高于截断值的恶性细胞定义为高代谢活性细胞,低于截断值的恶性细胞定义为低代谢活性细胞;
将降维聚类中恶性细胞以外的细胞定义为非恶性细胞。
进一步,所述定义代谢活性基因包括:
筛选转移群细胞和原发群细胞之间的差异基因;
对差异基因进行富集分析;
将代谢通路中富集的差异基因定义为代谢相关基因;
对代谢相关基因进行GSEA分析,将转移细胞群中富集的代谢相关基因定义为代谢活性基因。
进一步,所述评分体系具体包括:
利用反卷积算法根据高代谢活性细胞和低代谢活性细胞的基因表达图谱,计算所有RNA序列数据对应患者的高代谢活性细胞的比例,定义该比例为评分体系。
另一方面,本发明提供一种基于单细胞分析建立的用于评估头颈鳞癌预后的评分系统,包括:
样本获取模块,用于获取样本数据,样本数据包括头颈鳞癌单细胞数据和大队列RNA数据,大队列RNA数据包括头颈鳞癌患者的RNA序列数据和临床信息;
质量控制模块,用于对所述单细胞数据和大队列RNA数据进行质量控制;
聚类注释模块,用于对所述单细胞数据中的细胞进行聚类和注释;
定义模块,用于定义恶性细胞和代谢活性基因;
细胞分群模块,用于根据代谢活性将细胞分群;
评分模块,用于根据分群后的细胞代谢活性与头颈鳞癌的相关性,建立不同细胞群比例与头颈鳞癌预后的关系作为评分体系;
筛选模块,用于根据评分体系筛选代谢生物标志物;
对所述单细胞数据进行质量控制具体包括:
去除表达基因≤200的细胞及基因表达数≥2500的细胞;
去除双细胞;
去除表达在少于3个细胞中的基因;
去除线粒体基因;
合并数据集;
对所述单细胞数据中的细胞进行聚类和注释具体包括:
对所有细胞进行降维聚类,过滤掉每个亚群中核糖体基因表达超过35%的细胞;
寻找每一亚群中的高表达基因,并根据寻找出的高表达基因,特殊表达的基因和已报道过细胞标记基因对每一细胞亚群进行注释;
注释后的细胞分为上皮细胞、T细胞、B细胞、浆细胞、单核细胞、肥大细胞、成纤维细胞及恶性细胞等亚群。
进一步,所述定义模块包括恶性细胞定义模块和代谢活性基因定义模块;
恶性细胞定义模块用于:
取样本数据中的恶性细胞进行降维聚类,根据每一群中恶性细胞的来源进行分类:转移癌样本来源最多的细胞群定义为转移群,原发癌样本来源最多的细胞群定义为原发群,其余从转移癌和原发癌样本来源相当的恶性细胞群定义为混合群;
代谢活性基因定义模块用于:
筛选转移群细胞和原发群细胞之间的差异基因;
对差异基因进行富集分析;
将代谢通路中富集的差异基因定义为代谢相关基因;
对代谢相关基因进行GSEA分析,将转移细胞群中富集的代谢相关基因定义为代谢活性基因。
进一步,评分模块具体包括:
利用反卷积算法根据高代谢活性细胞和低代谢活性细胞的基因表达图谱,计算所有RNA序列数据对应患者的高代谢活性细胞的比例,定义该比例为评分体系。
本发明的有益效果为:
1.将单细胞测序技术和大队列RNA-Seq技术相结合,从细胞和临床预后两个层面基于代谢活性构建了HNSCC的评分系统。
2.为进一步分析HNSCC的代谢特征,以及代谢重编程如何影响HNSCC患者的肿瘤恶性程度和耐药性提供参考。
附图说明
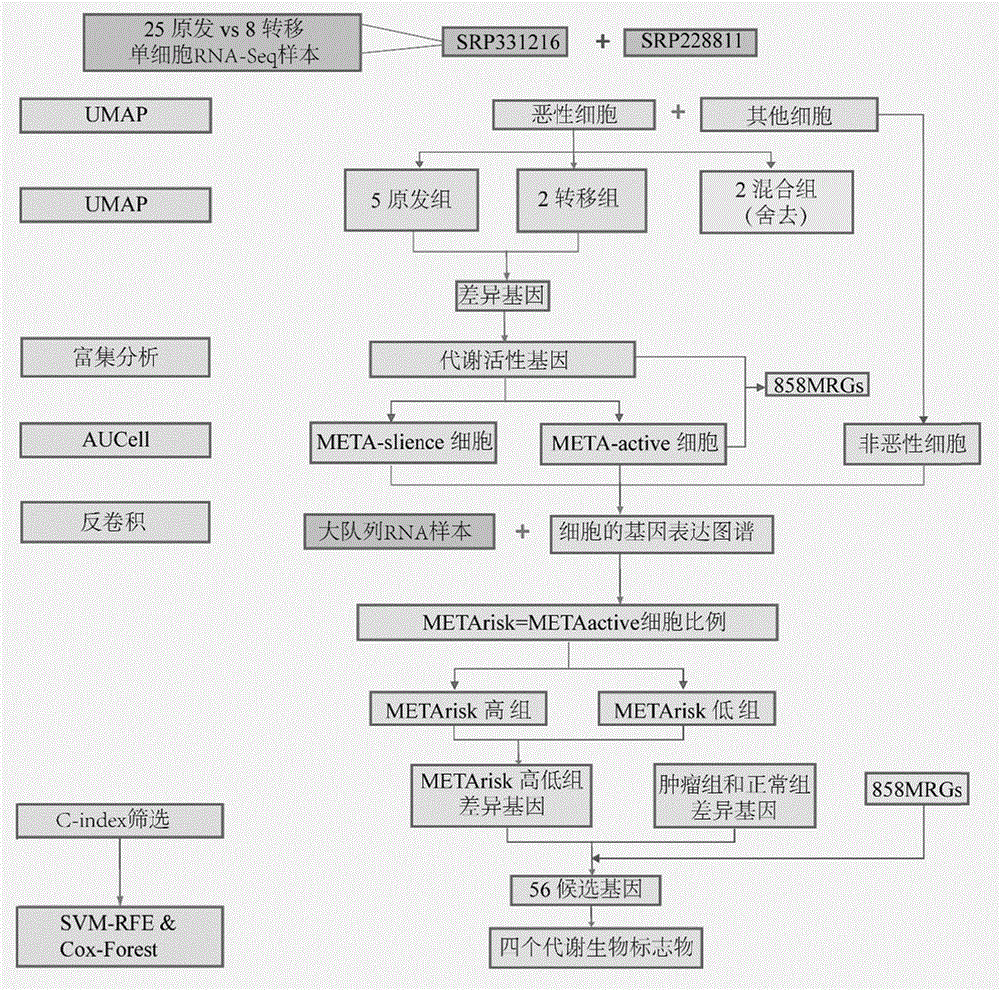
图1为本发明实施例一的方法流程图。
图2为本发明实施例一的METArisk评分体系的构建流程图。
图3为本发明实施例一的代谢生物标志物筛选效果图。
具体实施方式
为了使本申请的目的、技术方案和优点更加清楚,下面结合附图对本申请具体实施例作进一步的详细描述。可以理解的是,此处所描述的具体实施例仅仅用于解释本申请,而非对本申请的限定。另外还需要说明的是,为了便于描述,附图中仅示出了与本申请相关的部分而非全部内容。在更加详细地讨论示例性实施例之前应当提到的是,一些示例性实施例被描述成作为流程图描绘的处理或方法。虽然流程图将各项操作(或步骤)描述成顺序的处理,但是其中的许多操作可以被并行地、并发地或者同时实施。此外,各项操作的顺序可以被重新安排。当其操作完成时所述处理可以被终止,但是还可以具有未包括在附图中的附加步骤。所述处理可以对应于方法、函数、规程、子例程、子程序等等。
下面将结合本申请实施例中的附图,对本申请实施例中的技术方案进行清楚地描述,显然,所描述的实施例是本申请一部分实施例,而不是全部的实施例。基于本申请中的实施例,本领域普通技术人员获得的所有其他实施例,都属于本申请保护的范围。
本申请的说明书和权利要求书中的术语“第一”、“第二”等是用于区别类似的对象,而不用于描述特定的顺序或先后次序。应该理解这样使用的数据在适当情况下可以互换,以便本申请的实施例能够以除了在这里图示或描述的那些以外的顺序实施,且“第一”、“第二”等所区分的对象通常为一类,并不限定对象的个数,例如第一对象可以是一个,也可以是多个。此外,说明书以及权利要求中“和/或”表示所连接对象的至少其中之一,字符“/”,一般表示前后关联对象是一种“或”的关系。
下面结合附图,通过具体的实施例及其应用场景对本申请实施例提供的进行详细地说明。
实施例一
如图1所示,本实施例提供一种基于单细胞分析建立的用于评估头颈鳞癌预后的评分方法,具体包括:
1.获取公共数据:
(1)获取单细胞数据:从NCBI的SRA数据库中下载序列号为SRP332116,228811的HNSCC单细胞数据,其中包括25例原发癌组织和8例转移癌,并利用cellranger(版本4.0.0)获取原始数据中的fastq文件,通过R语言中的Seruat(版本4.0.4)和pipeline(版本4.0.5)获取基因barcode矩阵。
(2)获取大队列RNA数据:从TCGA和GEO数据库下载486例HNSCC患者的大队列RNA-Seq数据及临床信息
2.数据的处理:对单细胞数据进行质量控制:(1)去除表达基因数≤200的细胞及基因表达数≥2500的细胞;(2)去除双细胞;(3)去除表达在少于3个细胞中的基因;(4)去除线粒体基因;同时利用‘ScaleData’函数将表达矩阵转换为易于分析的格式,用‘FindIntegrationAchors’和‘IntegrateData’函数来合并不同数据集。对大队列RNA数据进行质量控制:(1)保留未被福尔马林浸泡的样本;(2)保留有完整临床数据的样本;(3)保留有明确临床结局及生存时间的样本
3.细胞聚类及注释:通过R语言中的Seurate包内的FindClusters函数对所有细胞进行降维聚类,过滤掉每个亚群中核糖体基因表达超过35%的细胞,并将聚类后的分群用UMAP图展示。如图2所示,利用FindAllMarker寻找每一亚群中的高表达基因,并根据寻找出的高表达基因,特殊表达的基因和已报道过细胞标记基因对每一细胞亚群进行注释(图2A)。注释后的细胞分为上皮细胞、T细胞、B细胞、浆细胞、单核细胞、肥大细胞、成纤维细胞及恶性细胞等亚群。
4.恶性细胞的定义:我们取样本中的恶性细胞,对其进行降维聚类,并根据每一群中恶性细胞的来源不同对其进行分类:从转移癌样本来源较多的细胞群定义为转移群,从原发癌样本来源多的细胞群定义为原发群,从转移癌和原发癌样本来源相当的细胞群定义为混合群(图2B)。
5.定义代谢活性基因:通过单细胞特有的方法Psedobulk筛选转移群细胞和原发群细胞之间的差异基因,利用差异基因进行KEGG-GO富集分析,发现差异基因在代谢通路中富集(图2C)。取这些差异基因定义为代谢相关基因(Metabolism related genes,MRG),并利用clusterProfiler R包进行GSEA分析,发现大部分代谢相关基因在转移细胞群中富集(图2D),于是取这一部分在转移细胞群中富集的基因,定义为META_ACTIVE基因。
6.根据代谢活性将细胞分群:利用AUCell算法分析META_ACTIVE基因在每一细胞中的激活情况,以此来表示细胞的代谢活性。通过“AUCell_exploreThresholds”函数计算代谢活性的截断值,高于截断值的恶性细胞为高代谢活性的细胞,我们将其定义为METAactive细胞,低于截断值的细胞为低代谢活性的细胞,我们将其定义为METAsilence细胞(图2E)。取降维聚类中恶性细胞之外的其他细胞,并通过2019年文献报道过的方法进行整合(1),定义为non-malignant细胞
7.确定细胞代谢分型的生物学意义:绘制METAactive细胞和METAsilence细胞的PCA图,并将其与样本来源的PCA图相对比,我们发现METAactive细胞多来自于转移癌样本,而METAsilence细胞多来自于原发癌样本,因此高代谢活性的METAactive细胞与肿瘤的恶性行为密切相关(图2F)。
8.根据特定细胞群与预后的关系建立METArisk评分:利用反卷积算法Cibersortx28,根据METAactive和METAsilence两群细胞的单细胞基因表达图谱,推断大队列RNA-Seq数据中每一位患者METAactive细胞占所有细胞的比例,并定义该比例为METArisk评分(图2G)。
9.验证METArisk评分对HNSCC患者预后评估能力:依据METArisk评分将HNSCC患者分为高/低METArisk两种亚型,对两种亚型的患者进行Kaplan-meier生存分析,结果显示,低METArisk患者预后较好,高METArisk患者预后较差(图2H),说明我们的代谢评分METArisk可以对患者的预后风险进行评估。
10.如图3所示,利用METArisk筛选代谢生物标志物:通过差异分析,我们选出了高低METArisk分组之间的差异基因,并将其与代谢相关基因和肿瘤与正常组的差异基因取交集,获得56个备选基因(图3A)。为筛选出能够独立预测头颈鳞癌预后的代谢生物标志物,我们探索了不同组合的生存分析模型的组合方式,通过计算每种组合的C-指数(图3B),我们最终选择了SVM-RFE(图3C)与多因素Cox PH(图3D)分析的组合,并从56个备选基因中筛选出EPHX3,FDCSP,FAM3B and PYGL四个基因作为研究备选的代谢生物标志物。
实施例二
本实施例提供一种基于单细胞分析建立的用于评估头颈鳞癌预后的评分系统,具体包括:
样本获取模块,用于获取样本数据,样本数据包括头颈鳞癌单细胞数据和大队列RNA数据,大队列RNA数据包括头颈鳞癌患者的RNA序列数据和临床信息;
质量控制模块,用于对所述单细胞数据和大队列RNA数据进行质量控制;
聚类注释模块,用于对所述单细胞数据中的细胞进行聚类和注释;
定义模块,用于定义恶性细胞和代谢活性基因;
细胞分群模块,用于根据代谢活性将细胞分群;
评分模块,用于根据分群后的细胞代谢活性与头颈鳞癌的相关性,建立不同细胞群比例与头颈鳞癌预后的关系作为评分体系;
筛选模块,用于根据评分体系筛选代谢生物标志物;
对所述单细胞数据进行质量控制具体包括:
去除表达基因≤200的细胞及基因表达数≥2500的细胞;
去除双细胞;
去除表达在少于3个细胞中的基因;
去除线粒体基因;
合并数据集;
对所述单细胞数据中的细胞进行聚类和注释具体包括:
对所有细胞进行降维聚类,过滤掉每个亚群中核糖体基因表达超过35%的细胞;
寻找每一亚群中的高表达基因,并根据寻找出的高表达基因,特殊表达的基因和已报道过细胞标记基因对每一细胞亚群进行注释;
注释后的细胞分为上皮细胞、T细胞、B细胞、浆细胞、单核细胞、肥大细胞、成纤维细胞及恶性细胞等亚群。
获取单细胞数据:从NCBI的SRA数据库中下载单细胞数据,并利用cellranger(版本4.0.0)获取原始数据中的fastq文件,通过R语言中的Seruat(版本4.0.4)和pipeline(版本4.0.5)获取基因barcode矩阵。
获取大队列RNA数据:从TCGA和GEO数据库下载HNSCC患者的大队列RNA-Seq数据及临床信息。
对单细胞数据进行质量控制:(1)去除表达基因数≤200的细胞及基因表达数≥2500的细胞;(2)去除双细胞;(3)去除表达在少于3个细胞中的基因;(4)去除线粒体基因;同时利用‘ScaleData’函数将表达矩阵转换为易于分析的格式,用‘FindIntegrationAchors’和‘IntegrateData’函数来合并不同数据集。对大队列RNA数据进行质量控制:(1)保留未被福尔马林浸泡的样本;(2)保留有完整临床数据的样本;(3)保留有明确临床结局及生存时间的样本。
细胞聚类及注释:通过R语言中的Seurate包内的FindClusters函数对所有细胞进行降维聚类,过滤掉每个亚群中核糖体基因表达超过35%的细胞,并将聚类后的分群用UMAP图展示。利用FindAllMarker寻找每一亚群中的高表达基因,并根据寻找出的高表达基因,特殊表达的基因和已报道过细胞标记基因对每一细胞亚群进行注释。注释后的细胞分为上皮细胞、T细胞、B细胞、浆细胞、单核细胞、肥大细胞、成纤维细胞及恶性细胞等亚群。
优选地,所述定义模块包括恶性细胞定义模块和代谢活性基因定义模块;
恶性细胞定义模块用于:
取样本数据中的恶性细胞进行降维聚类,根据每一群中恶性细胞的来源进行分类:
转移癌样本来源最多的细胞群定义为转移群,原发癌样本来源最多的细胞群定义为原发群,其余从转移癌和原发癌样本来源相当的恶性细胞群定义为混合群,相关代码如下:
/>
/>
/>
/>
/>
/>
代谢活性基因定义模块用于:
筛选转移群细胞和原发群细胞之间的差异基因;
对差异基因进行富集分析;
将代谢通路中富集的差异基因定义为代谢相关基因;
对代谢相关基因进行GSEA分析,将转移细胞群中富集的代谢相关基因定义为代谢活性基因,通过单细胞特有的方法Psedobulk筛选转移群细胞和原发群细胞之间的差异基因,用差异基因进行KEGG-GO富集分析,发现差异基因在代谢通路中富集。取这些差异基因定义为代谢相关基因(Metabolism related genes,MRG),并利用clusterProfiler R包进行GSEA分析,发现大部分代谢相关基因在转移细胞群中富集,于是取这一部分在转移细胞群中富集的基因,定义为META_ACTIVE基因。相关代码如下:
/>
/>
/>
/>
根据代谢活性将细胞分群:利用AUCell算法分析META_ACTIVE基因在每一细胞中的激活情况,以此来表示细胞的代谢活性。通过“AUCell_exploreThresholds”函数计算代谢活性的截断值,高于截断值的恶性细胞为高代谢活性的细胞,我们将其定义为METAactive细胞,低于截断值的细胞为低代谢活性的细胞,我们将其定义为METAsilence细胞。取降维聚类中恶性细胞之外的其他细胞,并通过首创的算法进行整合,定义为非恶性细胞,相关代码如下:
/>
/>
优选地,评分模块具体包括:
利用反卷积算法根据高代谢活性细胞和低代谢活性细胞的基因表达图谱,计算所有RNA序列数据对应患者的高代谢活性细胞的比例,定义该比例为评分体系。根据特定细胞群与预后的关系建立METArisk评分:利用反卷积算法,根据METAactive和METAsilence两群细胞的单细胞基因表达图谱,推断大队列RNA-Seq数据中每一位患者METAactive细胞占所有细胞的比例,并定义该比例为METArisk评分。相关代码如下:
/>
验证METArisk评分对HNSCC患者预后评估能力:依据METArisk评分将HNSCC患者分为高/低METArisk两种亚型,对两种亚型的患者进行Kaplan-meier生存分析,结果显示,低METArisk患者预后较好,高METArisk患者预后较差,说明我们的代谢评分METArisk可以对患者的预后风险进行评估。相关代码如下:
/>
利用METArisk筛选代谢生物标志物:通过差异分析,我们选出了高低METArisk分组之间的差异基因,并将其与代谢相关基因和肿瘤与正常组的差异基因取交集,获得56个备选基因。为筛选出能够独立预测头颈鳞癌预后的代谢生物标志物,我们探索了不同组合的生存分析模型的组合方式,通过计算每种组合的C-指数,我们最终选择了SVM-RFE与多因素Cox PH分析的组合,并从56个备选基因中筛选出EPHX3,FDCSP,FAM3B and PYGL四个基因。相关代码如下:
BiocManager::install("mixOmics")
BiocManager::install("CoxBoost")
library(devtools)
install_github("binderh/CoxBoost")
library(survival)
library(randomForestSRC)
library(glmnet)
library(plsRcox)
library(superpc)
library(gbm)
library(CoxBoost)
library(survivalsvm)
library(dplyr)
library(tibble)
library(BART)
library(limma)
library(tidyverse)
library(dplyr)
library(miscTools)
library(compareC)
library(ggplot2)
library(ggsci)
library(tidyr)
library(ggbreak)
Sys.setenv(LANGUAGE="en")#显示英文报错信息
options(stringsAsFactors=FALSE)#禁止chr转成factor
#加载数据集
#tcga<-read.table("TCGA.txt",header=T,sep=" ",quote="",check.names=F)
#GSE57303<-read.table("GSE57303.txt",header=T,sep=" ",quote="",check.names=F)
#GSE62254<-read.table("GSE62254.txt",header=T,sep=" ",quote="",check.names=F)
#生成包含三个数据集的列表
mm<-list(TCGA=tcga,
GSE65858=GSE65858,GSE41613=GSE41613)
#数据标准化
mm<-lapply(mm,function(x){
x[,-c(1:3)]<-scale(x[,-c(1:3)])
return(x)})
result<-data.frame()
#TCGA作为训练集
est_data<-mm$TCGA
#GEO作为验证集
val_data_list<-mm
pre_var<-colnames(est_data)[-c(1:3)]
est_dd<-est_data[,c('OS.time','OS',pre_var)]
val_dd_list<-lapply(val_data_list,function(x){x[,c('OS.time','OS',pre_var)]})
#设置种子数和节点数,其中节点数可以调整
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
#将得到的结果赋给result2变量进行操作
result2<-result
###将结果的长数据转换为宽数据
dd2<-pivot_wider(result2,names_from='ID',values_from='Cindex')%>%as.data.frame()
#将C指数定义为数值型
dd2[,-1]<-apply(dd2[,-1],2,as.numeric)
#求每个模型的C指数在三个数据集的均值
dd2$All<-apply(dd2[,2:4],1,mean)
#求每个模型的C指数在GEO验证集的均值
dd2$GEO<-apply(dd2[,3:4],1,mean)
###查看每个模型的C指数
head(dd2)
#输出C指数结果
#write.table(dd2,"output_C_index.txt",col.names=T,row.names=F,sep=" ",quote=F)
dd2=read.table('output_C_index.txt',header=T,sep=" ",check.names=F)
library(ComplexHeatmap)
library(circlize)
library(RColorBrewer)
#根据C指数排序
dd2<-dd2[order(dd2$GEO,decreasing=T),]
#仅绘制GEO验证集的C指数热图
dt<-dd2[,3:4]
rownames(dt)<-dd2$Model
##热图绘制
/>
需要说明的是,在本文中,所述存储介质,包括计算机可读存储介质,如计算机只读存储器(Read-Only Memory,ROM)、随机存取存储器(Random Access Memory,RAM)、磁碟或者光盘等。
术语“包括”、“包含”或者其任何其他变体意在涵盖非排他性的包含,从而使得包括一系列要素的过程、方法、物品或者装置不仅包括那些要素,而且还包括没有明确列出的其他要素,或者是还包括为这种过程、方法、物品或者装置所固有的要素。在没有更多限制的情况下,由语句“包括一个……”限定的要素,并不排除在包括该要素的过程、方法、物品或者装置中还存在另外的相同要素。此外,需要指出的是,本申请实施方式中的方法和装置的范围不限按示出或讨论的顺序来执行功能,还可包括根据所涉及的功能按基本同时的方式或按相反的顺序来执行功能,例如,可以按不同于所描述的次序来执行所描述的方法,并且还可以添加、省去、或组合各种步骤。另外,参照某些示例所描述的特征可在其他示例中被组合。
通过以上的实施方式的描述,本领域的技术人员可以清楚地了解到上述实施例方法可借助软件加必需的通用硬件平台的方式来实现,当然也可以通过硬件,但很多情况下前者是更佳的实施方式。基于这样的理解,本申请的技术方案本质上或者说对现有技术做出贡献的部分可以以计算机软件产品的形式体现出来,该计算机软件产品存储在一个存储介质(如ROM/RAM、磁碟、光盘)中,包括若干指令用以使得一台终端(可以是手机,计算机,服务器,或者网络设备等)执行本申请各个实施例所述的方法。
上面结合附图对本申请的实施例进行了描述,但是本申请并不局限于上述的具体实施方式,上述的具体实施方式仅仅是示意性的,而不是限制性的,本领域的普通技术人员在本申请的启示下,在不脱离本申请宗旨和权利要求所保护的范围情况下,还可做出很多形式,均属于本申请的保护之内。
上述仅为本申请的较佳实施例及所运用的技术原理。本申请不限于这里所述的特定实施例,对本领域技术人员来说能够进行的各种明显变化、重新调整及替代均不会脱离本申请的保护范围。因此,虽然通过以上实施例对本申请进行了较为详细的说明,但是本申请不仅仅限于以上实施例,在不脱离本申请构思的情况下,还可以包括更多其他等效实施例,而本申请的范围由权利要求的范围决定。
- 一种鳞癌的预后标志物、预后风险评估模型的建立方法及其应用
- 一种头颈鳞癌免疫联合化疗预后评估生物标志物及其应用