肿瘤分子分型方法及装置、终端设备及可读存储介质
文献发布时间:2023-06-19 10:54:12
技术领域
本发明涉及生物医学技术领域,尤其涉及一种肿瘤分子分型方法及装置、终端设备及可读存储介质。
背景技术
肿瘤是人体中正在发育或成熟的正常细胞,在某些致瘤因素的作用下某些细胞群出现过度增殖或者异常分化而生成的局部肿块。与正常组织细胞相比,这些细胞的新陈代谢不符合正常规律,也不会正常死亡。这种不受约束的生长导致细胞形态和功能异常,从而破坏了正常组织器官的结构并影响其功能。肿瘤是一种由于基因改变导致的疾病。内部或者外部的致瘤因素诱发体细胞基因突变,而失常的基因引发一系列异常生物学途径,从而改变细胞形态与功能。研究表明肿瘤的发生是多基因,多步骤的复杂疾病。恶性肿瘤(癌症)已经成为严重威胁中国人群健康的主要公共卫生问题之一,根据最新的统计数据显示,恶性肿瘤死亡占居民全部死因的23.91%,且近十几年来恶性肿瘤的发病死亡均呈持续上升态势,每年恶性肿瘤所致的医疗花费超过2200亿,防控形势严峻。为了提肿瘤的诊疗水平及治愈率,基于基因组大数据和机器学习算法对肿瘤进行分子分型来推动精准医疗显得尤为重要。
胰腺癌在中国是一种较为高发且难治的癌症,据2015年的癌症统计数据显示,胰腺癌的发病率在癌症领域排名第9,死亡率排名第6。以胰腺癌为例,虽然目前存在针对胰腺癌的分子分型的技术方案,但均存在各种缺陷,包括分子分型与预后关系不紧密、不同分子分型的基因组特征与癌变机制不明确、分子分型无法与特定的治疗方案建立联系等。例如,一类技术方案为基于同源重组修复基因(Homologous Recombination Repair, HRR)是否突变将胰腺癌进行分子分型,但是该技术方案只能区分应用铂类化疗或PARP(poly(ADP-ribose) polymerase)抑制剂治疗患者的预后,对于接受其他治疗方式的胰腺癌患者的预后无法有效区分,可见该分子分型技术方案存在较大的局限性。还有一类技术方案为基于基因组大片段扩增和缺失对胰腺癌患者进行分子分型,但该技术用于进行分子分型的数据分辨率过低,无法获得精准的分子分型,且该结果与患者预后并不具有显著的相关性。再有一类技术方案为基于肿瘤组织的转录谱对胰腺癌患者进行分子分型,但该技术对肿瘤组织的质量要求较高,否则无法获取准确的转录谱数据,限制了该技术的临床应用,且该技术并不能区分所有分型之间的预后差异。
发明内容
针对上述问题,本发明提供了一种肿瘤分子分型方法及装置、终端设备及可读存储介质,有效解决现有分子分型方法对肿瘤组织的质量要求较高、不能区分所有分型之间的预后差异等技术问题。
本发明提供的技术方案如下:
一方面,本发明提供了一种肿瘤分子分型方法,包括:
获取多个肿瘤组织样本的测序数据,并基于预先构建的覆盖深度基线计算各基因的拷贝数数值;
根据预设的拷贝数变异数阈值和计算得到的拷贝数数值筛选各肿瘤组织样本中发生变异的基因;
基于选定基因的拷贝数数值对肿瘤组织样本进行无监督聚类得到针对不同变异特征的多个样本类别;
筛选各样本类别样本间基因拷贝数变异达到预设差异的基因;
针对筛选得到的基因进一步进行无监督聚类得到针对不同变异特征的多个基因类别;
基于拷贝数变异分别计算各基因类别中基因的第一主成分,并通过回归分析的方法确定各基因类别第一主成分对患者预后的影响;
根据基因第一主成分对患者预后的影响计算各肿瘤组织样本的拷贝数变异分值CNV score,并根据无监督聚类结果和拷贝数变异分值CNV score进一步对各样本类别的样本进行分类,完成对肿瘤的分子分型;其中,拷贝数变异分值CNV score为:
其中,
进一步优选地,所述通过回归分析的方法确定各基因类别第一主成分对患者预后的影响中,包括:
分别对各基因类别中包含基因的第一主成分进行回归分析确定相关方向;
根据确定的相关方向确定回归系数方向;
根据回归系数的方向确定各基因类别中基因拷贝数变异的第一主成分对患者预后的影响;其中,当回归系数为负方向,表示相应基因利于患者预后;当回归系数为正方向,表示相应基因不利于患者预后。
进一步优选地,所述根据无监督聚类结果和拷贝数变异分值CNV score进一步对各样本类别的样本进行分类,完成对肿瘤的分子分型中,包括:
根据计算得到的拷贝数变异分值CNV score、通过遍历的方法选定基因拷贝数变异分值阈值,使得同一样本类别中不同分组的肿瘤组织样本对应患者的预后差异最大;
根据选定的基因拷贝数变异分值阈值分别对各样本类别中的肿瘤组织样本进行亚组细分,完成对肿瘤的分子分型。
进一步优选地,在所述基于选定基因的拷贝数数值对肿瘤组织样本进行无监督聚类得到针对不同变异特征的多个样本类别及在所述针对筛选得到的基因进一步进行无监督聚类得到针对不同变异特征的多个基因类别中,均采用PAM算法进行无监督聚类,并通过轮廓系数确定最优聚类数目;
和/或在所述筛选各样本类别样本间基因拷贝数变异达到预设差异的基因中,采用秩和检验的方法筛选各样本类别样本间基因拷贝数变异达到预设差异的基因,其中,当秩和检验p值小于预设阈值,确定相应基因达到预设差异。
进一步优选地,所述基于选定基因的拷贝数数值对肿瘤组织样本进行无监督聚类得到针对不同变异特征的多个样本类别中,包括:
对各选定基因的拷贝数数值进行z-score标准化处理;
基于计算得到z-score值对肿瘤组织样本进行无监督聚类得到针对不同变异特征的多个样本类别。
进一步优选地,所述获取多个肿瘤组织样本的测序数据,基于预先构建的覆盖深度基线得到各基因的拷贝数数值中,包括:
获取多个肿瘤组织样本的测序数据并对其进行预处理操作;
根据健康人的白细胞测序数据为每个基因捕获区域构建覆盖深度基线;
根据预处理操作后的肿瘤组织样本测序数据计算每个基因捕获区域的覆盖深度;
依次计算各基因捕获区域的拷贝数数值,其中,针对一基因捕获区域,基于该基因捕获区域在肿瘤组织样本中的覆盖深度和构建的覆盖深度基线比值的log2转化值计算得到拷贝数数值;
根据计算得到的基因捕获区域的拷贝数数值得到基因的拷贝数数值,其中,针对一基因,其拷贝数数值由该基因覆盖的所有基因捕获区域的拷贝数数值的集中趋势计算得到。
本发明还提供了一种肿瘤分子分型装置,包括:
基因拷贝数数值计算模块,用于获取多个肿瘤组织样本的测序数据,并基于预先构建的覆盖深度基线计算各基因的拷贝数数值;
变异基因筛选模块,用于根据预设的拷贝数变异数阈值和基因拷贝数数值计算模块计算得到的拷贝数数值筛选各肿瘤组织样本中发生变异的基因;
第一聚类模块,用于基于变异基因筛选模块选定基因的拷贝数数值对肿瘤组织样本进行无监督聚类得到针对不同变异特征的多个样本类别;
差异基因筛选模块,用于筛选第一聚类模块聚类后各样本类别样本间基因拷贝数变异达到预设差异的基因;
第二聚类模块,用于针对差异基因筛选模块筛选得到的基因进一步进行无监督聚类得到针对不同变异特征的多个基因类别;
预后影响分析模块,用于基于拷贝数变异分别计算第二聚类模块聚类后各基因类别中基因的第一主成分,并通过回归分析的方法确定各基因类别第一主成分对患者预后的影响;
样本分类模块,用于根据基因第一主成分对患者预后的影响计算各肿瘤组织样本的拷贝数变异分值CNV score,并根据无监督聚类结果和拷贝数变异分值CNV score进一步对各样本类别的样本进行分类,完成对肿瘤的分子分型;其中,拷贝数变异分值CNV score为:
其中,
进一步优选地,所述预后影响分析模块中包括:
回归单元,用于分别对各基因类别中包含基因的第一主成分进行回归分析确定相关方向;
回归系数确定单元,用于根据回归单元确定的相关方向确定回归系数方向;
患者预后确定单元,用于根据回归系数的方向确定各基因类别中基因拷贝数变异的第一主成分对患者预后的影响;其中,当回归系数为负方向,表示相应基因利于患者预后;当回归系数为正方向,表示相应基因不利于患者预后。
进一步优选地,所述样本分类模块中还包括:
阈值确定单元,用于根据计算得到的拷贝数变异分值CNV score、通过遍历的方法选定基因拷贝数变异分值阈值,使得同一样本类别中不同分组的肿瘤组织样本对应患者的预后差异最大;
分类单元,用于根据阈值确定单元选定的基因拷贝数变异分值阈值分别对各样本类别中的肿瘤组织样本进行亚组细分,完成对肿瘤的分子分型。
进一步优选地,所述第一聚类模块中包括:
标准化单元,用于对各选定基因的拷贝数数值进行z-score标准化处理;
第一聚类单元,用于基于标准化单元计算得到z-score值对肿瘤组织样本进行无监督聚类得到针对不同变异特征的多个样本类别。
进一步优选地,所述基因拷贝数数值计算模块中包括:
预处理单元,用于获取多个肿瘤组织样本的测序数据并对其进行预处理操作;
基线构建单元,用于根据健康人的白细胞测序数据为每个基因捕获区域构建覆盖深度基线;
覆盖深度计算单元,用于根据预处理单元预处理后的肿瘤组织样本测序数据计算每个基因捕获区域的覆盖深度;
拷贝数数值计算单元,用于依次计算各基因捕获区域的拷贝数数值,其中,针对一基因捕获区域,基于该基因捕获区域在肿瘤组织样本中的覆盖深度和构建的覆盖深度基线比值的log2转化值计算得到拷贝数数值;
基因拷贝数数值确定单元,用于根据拷贝数数值计算单元计算得到的基因捕获区域的拷贝数数值得到基因的拷贝数数值,其中,针对一基因,其拷贝数数值由该基因覆盖的所有基因捕获区域的拷贝数数值的集中趋势计算得到。
另一方面,本发明还提供了一种终端设备,包括存储器、处理器以及存储在所述存储器中并可在所述处理器上运行的计算机程序,所述处理器运行所述计算机程序时实现上述肿瘤分子分型方法的步骤。
另一方面,一种计算机可读存储介质,所述计算机可读存储介质存储有计算机程序,所述计算机程序被处理器执行时实现上述肿瘤分子分型方法的步骤。
本发明提供的肿瘤分子分型方法及装置、终端设备及可读存储介质中,至少能够带来以下有益效果:
1.该肿瘤分子分型方法和装置基于中国人基因组数据开发,可以非常有效地区分中国肿瘤患者(尤其是胰腺癌患者)的预后差异,甚至可以直接反映与分子分型结果对应的癌变机制,供临床医生匹配药物和精准选择治疗方案做参考。
2.该肿瘤分子分型方法和装置不仅适用于接受过铂类化疗或PARP抑制剂治疗的患者进行分子分型,对接受不同治疗方案的肿瘤癌患者均适用,具有宽广的临床应用范围。
3.该肿瘤分子分型方法和装置基于每个基因的拷贝数变异进行分子分型,分辨率高、分型精准,可以显著区分不同分子分型肿瘤患者的预后,具有更高的临床应用价值。
4.该肿瘤分子分型方法和装置基于肿瘤组织DNA样本测序数据获取原始数据,降低了对样本质量的要求,且有潜力推广到基于患者体液样本DNA测序的应用场景,具有更高的临床应用可能性。
附图说明
下面将以明确易懂的方式,结合附图说明优选实施方式,对上述特性、技术特征、优点及其实现方式予以进一步说明。
图1为本发明中肿瘤分子分型方法流程示意图;
图2为本发明中肿瘤分子分型装置结构示意图;
图3为本发明中终端设备结构示意图。
附图标记:
100-肿瘤分子分型装置,110-基因拷贝数数值计算模块,120-变异基因筛选模块,130-第一聚类模块,140-差异基因筛选模块,150-第二聚类模块,160-预后影响分析模块,170-样本分类模块。
具体实施方式
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对照附图说明本发明的具体实施方式。显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图,并获得其他的实施方式。
本发明提供的第一种实施例,
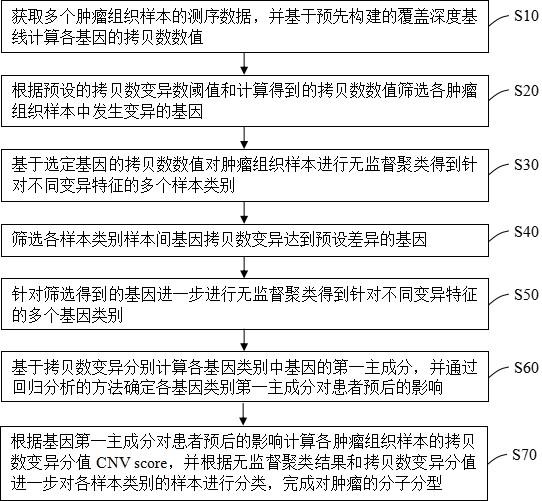
S10 获取多个肿瘤组织样本
S20 根据预设的拷贝数变异数阈值和计算得到的拷贝数数值筛选各肿瘤组织样本中发生变异的基因;
S30 基于选定基因的拷贝数数值对肿瘤组织样本进行无监督聚类得到针对不同变异特征的多个样本类别;
S40 筛选各样本类别样本间基因拷贝数变异达到预设差异的基因;
S50 针对筛选得到的基因进一步进行无监督聚类得到针对不同变异特征的多个基因类别;
S60 基于拷贝数变异分别计算各基因类别中基因的第一主成分,并通过回归分析的方法确定各基因类别第一主成分对患者预后的影响;
S70 根据基因第一主成分对患者预后的影响计算各肿瘤组织样本的拷贝数变异分值CNV score,并根据无监督聚类结果和拷贝数变异分值CNV score进一步对各样本类别的样本进行分类,完成对肿瘤的分子分型。在本实施例中,肿瘤组织样本的测序数据(FASTQ文件)为原始数据,在获取了原始的肿瘤组织样本测序数据之后,对其进行预处理操作即可对其进行分子分型操作,其中,预操作处理包括过滤、排序、重叠区域标记等,过滤包括对测序接头序列及低质量碱基的移除等。
一实例中,采用软件Trimmomatic(v0.36)对肿瘤组织样本的测序数据进行过滤, 将测序接头序列滤除之外,同时将符合以下条件的碱基移除:1.首端碱基质量小于3的碱 基;2.尾端碱基质量小于3的碱基;3.从5'端开始进行滑动,滑动位点周围4个碱基的范围内 碱基的平均碱基低于20处的碱基。之后,采用软件BWA(v0.7.17)将过滤后的测序数据回贴 至hg19版本的人类基因组上。接着,对回贴结果进行处理,包括采用软件Picard(v2.23.0) 基于依赖基因组坐标对回贴结果进行排序及对回贴结果中的重叠区域进行标记,完成对测 序
预处理操作完成之后,进入对基因计算拷贝数数值的步骤。在这一过程中,首先根据健康人的白细胞测序数据为每个基因捕获区域构建覆盖深度基线,并根据预处理操作后的肿瘤组织样本测序数据计算每个基因捕获区域的覆盖深度;之后,根据构建的覆盖深度基线和计算得到的肿瘤组织样本测序数据中基因捕获区域的覆盖深度计算相应基因捕获区域的拷贝数数值,其中,单个基因捕获区域的拷贝数数值由该基因捕获区域在肿瘤组织样本中的覆盖深度和构建的覆盖深度基线比值的log2转化值计算得到;最后,根据计算得到的基因捕获区域的拷贝数数值得到基因的拷贝数数值,其中,单个基因的拷贝数数值由其基因覆盖的所有基因捕获区域的拷贝数数值的集中趋势计算得到。
确定单个基因的拷贝数数值时,其覆盖范围内基因捕获区域拷贝数数值的集中趋势可以由多种方法得到,如,将多个拷贝数数值的中位数作为基因的拷贝数数值;又如,将多个拷贝数数值的平均值作为基因的拷贝数数值等。在实际应用中,可以根据实际情况灵活选用。另外,实例中可选用软件CNVkit(v0.9.2)对基因的拷贝数变异进行鉴定,也可以用其他适用软件。
对于基因拷贝数变异,通常认为变异类型包括肿瘤组织样本中发生基因缺失、基因扩增等,以此为了提高后续无监督聚类的精度,在得到基因的拷贝数数值之后,进一步根据预先设定的拷贝数变异数阈值将可能发生变异的基因筛选出来,其中,拷贝数变异数阈值由基因中可能发生的变异类型确定,如一实例中,将变异类型确定为基因缺失和基因扩增两类,并将拷贝数数值小于等于0.6的基因认为在肿瘤组织样本中发生缺失,将拷贝数数值大于等于1.6的基因认为在肿瘤组织样本中发生扩增。我们知道,对于不同种类的肿瘤组织样本,拷贝数变异数阈值均有可能发生变化,是以拷贝数变异数阈值同样需要根据实际情况进行选定,如,在另一实例中,将拷贝数数值小于等于0.8的基因认为在肿瘤组织样本中发生缺失,将拷贝数数值大于等于1.2的基因认为在肿瘤组织样本中发生扩增等。
完成对拷贝数变异基因的选定之后,随即对各选定基因的拷贝数数值进行z-score标准化处理,计算z-score值;进而基于计算得到z-score值对肿瘤组织样本进行无监督聚类得到针对不同变异特征的多个样本类别。由于采用无监督方法进行聚类,聚类效果(包括样本类别的数量等)与选用的聚类算法有直接关联,故根据应用需求对聚类算法进行选定,以聚类得到符合实际需求的样本类别。如,一实例中,选用的肿瘤组织样本为胰腺癌组织样本,采用PAM(partitioning around medoids)算法对由拷贝数数值计算得到的z-score值进行无监督聚类将胰腺癌组织样本分为两大类,过程中通过轮廓系数(silhouettemethod)确定最优聚类数目,且样本类别之间的差距通过计算欧式距离进行衡量。在其他实例中,还可以选用SOM(Self Organized Maps)、CLARA(Clustering LARge Applications)等方法进行无监督聚类,只要选用的聚类算法能够将样本聚类为需求类别即可。要说明的是,这一过程中针对基因的变异特征对肿瘤组织样本进行聚类,而不是变异类型,是以聚类后不同类别中的一个样本中可能存在不同类型的变异。
将肿瘤组织样本聚类为不同的样本类别之后,进一步通过秩和检验的方法筛选各样本类别样本间基因拷贝数变异达到预设差异的基因,并针对筛选得到的基因进一步进行无监督聚类得到针对不同变异特征的多个基因类别。具体,当无监督聚类得到的样本类别为两类,则使用Wilcoxon秩和检验方法对样本间基因拷贝数变异达到预设差异的基因进行鉴定;当无监督聚类得到的样本类别为两类以上,则使用Kruskal-Wallis检验方法(用于确定两个或多个基因组的中位数是否存在差异)对样本间基因拷贝数变异达到预设差异的基因进行鉴定,且当秩和检验p值小于预设阈值(通常设定为0.05),确定相应基因达到预设差异。与类别无监督聚类类似,基因类别聚类过程中使用的无监督算法同样可以根据实际情况进行选定,如,同样采用PAM算法进行无监督聚类将变异差异显著的基因聚为2类。
对达到预设差异的基因进行无监督聚类之后,基于拷贝数变异分别计算各基因类别中基因的第一主成分,并通过回归分析的方法确定各基因类别第一主成分对患者预后的影响。具体来说,分别对各基因类别中包含基因的第一主成分进行回归分析确定相关方向,进而根据确定的相关方向进一步确定回归系数方向,最后根据回归系数的方向确定各基因类别中基因拷贝数变异的第一主成分对患者预后的影响;其中,当回归系数为负方向,表示相应基因利于患者预后;当回归系数为正方向,表示相应基因不利于患者预后。回归算法可以选用Cox回归算法(Cox Regression)等,回归得到各类别基因的生存周期,包括总生存期OS、无进展生存期PFS等。
确定了各基因类别第一主成分对患者预后的影响之后,根据基因第一主成分对患者预后的影响计算各肿瘤组织样本的拷贝数变异分值CNV score,并根据无监督聚类结果和拷贝数变异分值CNV score进一步对各样本类别的样本进行分类,完成对肿瘤的分子分型。拷贝数变异分值CNV score如式(1):
其中,
之后,根据计算得到的拷贝数变异分值CNV score、通过遍历的方法选定基因拷贝数变异分值阈值,将同一样本类别中的肿瘤组织样本再一次分组,使得分组后不同组之间的肿瘤组织样本对应患者的预后差异最大,即由拷贝数变异分值得到的不同组别的生存曲线间隔最远。最后根据选定的基因拷贝数变异分值阈值分别对各样本类别中的肿瘤组织样本进行亚组细分,完成对肿瘤的分子分型。其中,阈值选定方法可以为最大选择检验方法(maximally selected rank statistics)等。
在一实例中,选用608个中国胰腺癌患者的组织样本进行分子分型,将变异类型确定为基因缺失和基因扩增两类,并将拷贝数数值小于等于0.6的基因认为在胰腺癌组织样本中发生缺失,将拷贝数数值大于等于1.6的基因认为在胰腺癌组织样本中发生扩增,筛选得到各肿瘤组织样本中发生变异的基因。之后,为筛选到的每个基因的拷贝数数值计算z-score,并基于PAM算法对胰腺癌组织样本进行无监督聚类得到两类,经分析,两类样本中的HRR基因分别对应扩增和非扩增,故将两类样本命名为HRR基因扩增组和HRR基因非扩增组。之后,通过Wilcoxon秩和检验方法鉴定出HRR基因扩增组和HRR基因非扩增组中p值小于0.05的基因,并使用PAM算法对其进行无监督聚类分为group1组和group2组。接着,基于PCA(主成分分析)算法分别为两组基因的拷贝数变异计算第一主成分PC1,并通过Cox回归分析确定两组基因拷贝数变异的PC1对患者预后的影响。之后,根据不同组别基因拷贝数变异的PC1和公式(1)分别计算每个胰腺癌组织样本的拷贝数变异分值CNV score,并通过最大选择检验方法(maximally selected rank statistics)选定基因拷贝数变异分值阈值,将HRR基因扩增组和HRR基因非扩增组进行亚组细分,获得4类分子分型,每类分子分型的患者数量如表1:
表1:608个胰腺癌组织样本分子分型结果
其中,拷贝数变异分值CNV score高表示该组别中胰腺癌组织样本的拷贝数变异分值CNV score大于基因拷贝数变异分值阈值,拷贝数变异分值CNV score低表示该组别中胰腺癌组织样本的拷贝数变异分值CNV score不大于基因拷贝数变异分值阈值。实践表明,分类后的4类分子分型之间预后差异显著,具有不同的胰腺癌基因组特征并可以预示可能适用的治疗方式,对治疗进行辅助。经分析,根据各类分子分型的特征,分别命名为:修复缺陷型(repair deficient)、增殖活跃型(proliferation active)、修复增强型(repairstrengthened)和修复超强型(repair ultra-strengthened),其中,修复缺陷型对应表1中HRR基因非扩增组中CNV score低的组别,增殖活跃型对应表1中HRR基因非扩增组中CNVscore高的组别,修复增强型对应表1中HRR基因扩增组中CNV score低的组别和修复超强型对应表1中HRR基因扩增组中CNV score高的组别。
本发明提供了一种肿瘤分子分型装置,如图2所示,该肿瘤分子分型装置100中包括:基因拷贝数数值计算模块110,用于获取多个肿瘤组织样本的测序数据,并基于预先构建的覆盖深度基线计算各基因的拷贝数数值;变异基因筛选模块120,用于根据预设的拷贝数变异数阈值和基因拷贝数数值计算模块计算得到的拷贝数数值筛选各肿瘤组织样本中发生变异的基因;第一聚类模块130,用于基于变异基因筛选模块选定基因的拷贝数数值对肿瘤组织样本进行无监督聚类得到针对不同变异特征的多个样本类别;差异基因筛选模块140,用于筛选第一聚类模块聚类后各样本类别样本间基因拷贝数变异达到预设差异的基因;第二聚类模块150,用于针对差异基因筛选模块筛选得到的基因进一步进行无监督聚类得到针对不同变异特征的多个基因类别;预后影响分析模块160,用于基于拷贝数变异分别计算第二聚类模块聚类后各基因类别中基因的第一主成分,并通过回归分析的方法确定各基因类别第一主成分对患者预后的影响;样本分类模块170,用于根据基因第一主成分对患者预后的影响计算各肿瘤组织样本的拷贝数变异分值CNV score,并根据无监督聚类结果和拷贝数变异分值CNV score进一步对各样本类别的样本进行分类,完成对肿瘤的分子分型。
在本实施例中,基因拷贝数数值计算模块110中包括:预处理单元、基线构建单元、覆盖深度计算单元、拷贝数数值计算单元及基因拷贝数数值确定单元。
肿瘤组织样本的测序数据(FASTQ文件)为原始数据,获取了原始的肿瘤组织样本测序数据之后,通过预处理单元对其进行预处理操作即可对其进行分子分型操作,其中,预操作处理包括过滤、排序、重叠区域标记等,过滤包括对测序接头序列及低质量碱基的移除等。之后,计算基因的拷贝数数值,包括:基线构建单元根据健康人的白细胞测序数据为每个基因捕获区域构建覆盖深度基线;覆盖深度计算单元根据健康人的白细胞测序数据为每个基因捕获区域构建覆盖深度基线,并根据预处理操作后的肿瘤组织样本测序数据计算每个基因捕获区域的覆盖深度;拷贝数数值计算单元根据构建的覆盖深度基线和计算得到的肿瘤组织样本测序数据中基因捕获区域的覆盖深度计算相应基因捕获区域的拷贝数数值,其中,单个基因捕获区域的拷贝数数值由该基因捕获区域在肿瘤组织样本中的覆盖深度和构建的覆盖深度基线比值的log2转化值计算得到;基因拷贝数数值确定单元根据计算得到的基因捕获区域的拷贝数数值得到基因的拷贝数数值,其中,单个基因的拷贝数数值由其基因覆盖的所有基因捕获区域的拷贝数数值的集中趋势计算得到。
确定单个基因的拷贝数数值时,其覆盖范围内基因捕获区域拷贝数数值的集中趋势可以由多种方法得到,如,将多个拷贝数数值的中位数作为基因的拷贝数数值;又如,将多个拷贝数数值的平均值作为基因的拷贝数数值等。在实际应用中,可以根据实际情况灵活选用。
对于基因拷贝数变异,通常认为变异类型包括肿瘤组织样本中发生基因缺失、基因扩增等,以此为了提高后续无监督聚类的精度,在得到基因的拷贝数数值之后,进一步根据预先设定的拷贝数变异数阈值将可能发生变异的基因筛选出来,其中,拷贝数变异数阈值由基因中可能发生的变异类型确定,如一实例中,将变异类型确定为基因缺失和基因扩增两类,并将拷贝数数值小于等于0.6的基因认为在肿瘤组织样本中发生缺失,将拷贝数数值大于等于1.6的基因认为在肿瘤组织样本中发生扩增。我们知道,对于不同种类的肿瘤组织样本,拷贝数变异数阈值均有可能发生变化,是以拷贝数变异数阈值同样需要根据实际情况进行选定,如,在另一实例中,将拷贝数数值小于等于0.8的基因认为在肿瘤组织样本中发生缺失,将拷贝数数值大于等于1.2的基因认为在肿瘤组织样本中发生扩增等。
第一聚类模块130中包括标准化单元及第一聚类单元。完成对拷贝数变异基因的选定之后,随即标准化单元对各选定基因的拷贝数数值进行z-score标准化处理,计算z-score值;进而第一聚类单元基于计算得到z-score值对肿瘤组织样本进行无监督聚类得到针对不同变异特征的多个样本类别。由于采用无监督方法进行聚类,聚类效果(包括样本类别的数量等)与选用的聚类算法有直接关联,故根据应用需求对聚类算法进行选定,以聚类得到符合实际需求的样本类别。如,一实例中,选用的肿瘤组织样本为胰腺癌组织样本,采用PAM算法对由拷贝数数值计算得到的z-score值进行无监督聚类将胰腺癌组织样本分为基因缺失组和基因非缺失组两大类,过程中通过轮廓系数(silhouette method)确定最优聚类数目,且样本类别之间的差距通过计算欧式距离进行衡量。在其他实例中,还可以选用SOM、CLARA等方法进行无监督聚类,只要选用的聚类算法能够将样本聚类为需求类别即可。
将肿瘤组织样本聚类为不同的样本类别之后,差异基因筛选模块140进一步通过秩和检验的方法筛选各样本类别样本间基因拷贝数变异达到预设差异的基因,并通过第二聚类模块150将筛选得到的基因进一步进行无监督聚类得到针对不同变异特征的多个基因类别。具体,当无监督聚类得到的样本类别为两类,则使用Wilcoxon秩和检验方法对样本间基因拷贝数变异达到预设差异的基因进行鉴定;当无监督聚类得到的样本类别为两类以上,则使用Kruskal-Wallis检验方法(用于确定两个或多个基因组的中位数是否存在差异)对样本间基因拷贝数变异达到预设差异的基因进行鉴定,且当秩和检验p值小于预设阈值(通常选定为0.05),确定相应基因达到预设差异。与类别无监督聚类类似,基因类别聚类过程中使用的无监督算法同样可以根据实际情况进行选定,如,同样采用PAM算法进行无监督聚类将变异差异显著的基因聚为2类。
预后影响分析模块160中包括:回归单元、回归系数确定单元及患者预后确定单元。对达到预设差异的基因进行无监督聚类之后,回归单元分别对各基因类别中包含基因的第一主成分进行回归分析确定相关方向,进而回归系数确定单元根据确定的相关方向进一步确定回归系数方向,最后患者预后确定单元根据回归系数的方向确定各基因类别中基因拷贝数变异的第一主成分对患者预后的影响;其中,其中,当回归系数为负方向,表示相应基因利于患者预后;当回归系数为正方向,表示相应基因不利于患者预后。回归算法可以选用Cox回归算法(Cox Regression)等,回归得到各类别基因的生存周期,包括总生存期OS、无进展生存期PFS等。
样本分类模块170中包括阈值确定单元及分类单元,其中,阈值确定单元根据计算得到的拷贝数变异分值、通过遍历的方法选定基因拷贝数变异分值阈值,将同一样本类别中的肿瘤组织样本再一次分组,使得分组后不同组之间的肿瘤组织样本对应患者的预后差异最大,即不同组别的生存曲线间隔最远。最后分类单元根据选定的基因拷贝数变异分值阈值对各样本类别中的肿瘤组织样本进行亚组细分,完成对肿瘤的分子分型。其中,阈值选定方法可以为最大选择检验方法(maximally selected rank statistics)等。
所属领域的技术人员可以清楚地了解到,为了描述的方便和简洁,仅以上述各程序模块的划分进行举例说明,实际应用中,可以根据需要而将上述功能分配由不同的程序模块完成,即将装置的内部结构划分成不同的程序单元或模块,以完成以上描述的全部或者部分功能。实施例中的各程序模块可以集成在一个处理单元中,也可是各个单元单独物理存在,也可以两个或两个以上单元集成在一个处理单元中,上述集成的单元既可以采用硬件的形式实现,也可以采用软件程序单元的形式实现。另外,各程序模块的具体名称也只是为了便于相互区分,并不用于限制本申请的保护范围。
图3是本发明一个实施例中提供的终端设备的结构示意图,如所示,该终端设备200包括:处理器220、存储器210以及存储在存储器210中并可在处理器220上运行的计算机程序211,例如:肿瘤分子分型关联程序。处理器220执行计算机程序211时实现上述各个肿瘤分子分型方法实施例中的步骤,或者,处理器220执行计算机程序211时实现上述肿瘤分子分型装置实施例中各模块的功能。
终端设备200可以为笔记本、掌上电脑、平板型计算机、手机等设备。终端设备200可包括,但不仅限于处理器220、存储器210。本领域技术人员可以理解,图3仅仅是终端设备200的示例,并不构成对终端设备200的限定,可以包括比图示更多或更少的部件,或者组合某些部件,或者不同的部件,例如:终端设备200还可以包括输入输出设备、显示设备、网络接入设备、总线等。
处理器220可以是中央处理单元(Central Processing Unit,CPU),还可以是其他通用处理器、数字信号处理器 (Digital Signal Processor,DSP)、专用集成电路(Application Specific Integrated Circuit,ASIC)、现场可编程门阵列(Field-Programmable Gate Array,FPGA)或者其他可编程逻辑器件、分立门或者晶体管逻辑器件、分立硬件组件等。通用处理器220可以是微处理器或者该处理器也可以是任何常规的处理器等。
存储器210可以是终端设备200的内部存储单元,例如:终端设备200的硬盘或内存。存储器210也可以是终端设备200的外部存储设备,例如:终端设备200上配备的插接式硬盘,智能TF存储卡(Smart Media Card,SMC),安全数字(Secure Digital,SD)卡,闪存卡(Flash Card)等。进一步地,存储器210还可以既包括终端设备200的内部存储单元也包括外部存储设备。存储器210用于存储计算机程序211以及终端设备200所需要的其他程序和数据。存储器210还可以用于暂时地存储已经输出或者将要输出的数据。
在上述实施例中,对各个实施例的描述都各有侧重,某个实施例中没有详细描述或记载的部分,可以参见其他实施例的相关描述。
本领域普通技术人员可以意识到,结合本文中所公开的实施例描述的各示例的单元及算法步骤,能够以电子硬件、或者计算机软件和电子硬件的结合来实现。这些功能究竟以硬件还是软件来执行,取决于技术方案的特定应用和设计约束条件。专业技术人员可以对每个特定的应用来使用不同方法来实现所描述的功能,但是这种实现不应认为超出本申请的范围。
在本申请所提供的实施例中,应该理解到,所揭露的装置/终端设备和方法,可以通过其他的方式实现。例如,以上所描述的装置/终端设备实施例仅仅是示意性的,例如,模块或单元的划分,仅仅为一种逻辑功能划分,实际实现时可以有另外的划分方式,例如,多个单元或组件可以结合或者可以集成到另一个系统,或一些特征可以忽略,或不执行。另一点,所显示或讨论的相互之间的耦合或直接耦合或通讯连接可以是通过一些接口,装置或单元的间接耦合或通讯连接,可以是电性、机械或其他的形式。
作为分离部件说明的单元可以是或者也可以不是物理上分开的,作为单元显示的部件可以是或者也可以不是物理单元,即可以位于一个地方,或者也可以分布到多个网络单元上。可以根据实际的需要选择其中的部分或者全部单元来实现本实施例方案的目的。
另外,在本申请各个实施例中的各功能单元可能集成在一个处理单元中,也可以是各个单元单独物理存在,也可以两个或两个以上单元集成在一个单元中。上述集成的单元既可以采用硬件的形式实现,也可以采用软件功能单元的形式实现。
集成的模块/单元如果以软件功能单元的形式实现并作为独立的产品销售或使用时,可以存储在一个计算机可读存储介质中。基于这样的理解,本发明实现上述实施例方法中的全部或部分流程,也可以通过计算机程序211发送指令给相关的硬件完成,的计算机程序211可存储于一计算机可读存储介质中,该计算机程序211在被处理器220执行时,可实现上述各个方法实施例的步骤。其中,计算机程序211包括:计算机程序代码,计算机程序代码可以为源代码形式、对象代码形式、可执行文件或某些中间形式等。计算机可读存储介质可以包括:能够携带计算机程序211代码的任何实体或装置、记录介质、U盘、移动硬盘、磁碟、光盘、计算机存储器、只读存储器 (ROM,Read-Only Memory)、随机存取存储器(RAM,RandomAccess Memory)、电载波信号、电信信号以及软件分发介质等。需要说明的是,计算机可读存储介质包含的内容可以根据司法管辖区内立法和专利实践的要求进行适当的增减,例如:在某些司法管辖区,根据立法和专利实践,计算机可读介质不包括电载波信号和电信信号。
应当说明的是,上述实施例均可根据需要自由组合。以上仅是本发明的优选实施方式,应当指出,对于本技术领域的普通相关人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 肿瘤分子分型方法及装置、终端设备及可读存储介质
- 膀胱癌分子分型方法及其装置、计算机可读存储介质