一种测定卡马西平和10-羟卡马西平的方法、试剂盒及其应用
文献发布时间:2023-06-19 11:22:42
技术领域
本发明属于医药检测技术领域,具体涉及一种测定卡马西平和10-羟卡马西平的方法、试剂盒及其应用。
背景技术
公开该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
癫痫是大脑神经元细胞异常放电引起的功能异常性疾病,该病由多种原因诱发,发作时呈短暂、突然、反复的特点,给患者及家属带来沉重身心痛苦及医疗负担。一般来讲,确诊癫痫的患者需持续规律服用抗癫痫药物,以减轻因癫痫反复发作对认知功能的侵害。卡马西平和奥卡西平作为临床常用抗癫痫药物,其联用治疗常用于难治性癫痫。卡马西平的抗惊厥机制尚不清楚,它可能能够增加钠通道的灭活功效,限制突触后神经元并阻断突触前Na
关于使用高效液相色谱-紫外色谱法(HPLC-UV)、气相色谱法(GC)和高效液相色谱-质谱联用法(HPLC-MS/MS)对生物液中抗癫痫药物进行定量的论文已经发表。刘勋等人在2020年采用HPLC-UV方法建立了同时检测4种抗癫痫药物的含量测定方法,但分析时间40分钟太长,无法实现高通量。代静等人报道了首次采用HPLC-MS/MS测定血清中12种抗癫痫药物的方法,方法检测了卡马西平和奥卡西平,并没有检测10-羟卡马西平,其原因是卡马西平和10-羟卡马西平性质相差较小,很难在普通C18色谱柱上得到有效分离。庄奕筠等人报道开发了沉淀蛋白法结合超高效液相色谱方法,该方法使用10μL样品体积,卡马西平和10-羟卡马西平的最低定量限(LLOQ)仅为1μg/mL和2.5μg/mL,其高进样量并不能得到理想的定量限,给临床药物浓度检测造成困难,且总分析时间长达24分钟,色谱峰拖尾且基线较高。因此,在药代动力学研究中,提出一种同时测定卡马西平和奥卡西平活性代谢物10-羟卡马西平含量的高灵敏度、高分离度的方法亟待解决。
发明内容
针对现有技术的不足,本发明提供一种测定卡马西平和10-羟卡马西平的方法、试剂盒及其应用。本发明采用沉淀蛋白法结合HPLC-MS/MS测定抗癫痫药物卡马西平和10-羟卡马西平在人体血浆中的含量,并从特异性、线性、灵敏度、准确度、精密度、基质效应、回收率和稳定性等方面对卡马西平和10-羟卡马西平在人血浆中的测定方法进行了充分的验证,最终应用于口服给药的人体药动学研究,并能够通过非房室模型计算药动学参数,因此具有良好的实际应用之价值。
为实现上述技术目的,本发明采用的技术方案如下:
本发明的第一个方面,提供一种测定卡马西平和10-羟卡马西平的方法,所述方法包括:以标准品制作标准曲线定量,同时采用质控品进行质控,以及基于HPLC-MS/MS对待测样品进行检测;
具体的,采用定量下限、低、中和高四个水平的质控品进行质控或采用低、中和高三个水平的质控品进行质控。
其中,所述卡马西平和10-羟卡马西平质控品的定量下限、低、中和高浓度均分别为0.1、0.25、2.5和25ng/mL。
所述待测样品的配制方法为:将试验样品与内标工作液混合,离心取上清制得。
所述试验样品为受试者血液样品,包括全血、血浆或血清,进一步优选为血浆。
所述内标工作液为同位素内标的卡马西平(卡马西平-d10)和10-羟卡马西平(10-羟卡马西平-d3)溶液,具体制备方法包括:将同位素内标的卡马西平和10-羟卡马西平原料药用乙腈溶解,制备内标储备液,然后使用沉淀蛋白溶剂稀释。
所述沉淀蛋白溶剂为乙腈与甲醇的混合溶液,所述乙腈和甲醇的用量体积比为2~4:1~3,优选为3:2。在此用量体积比下,其沉淀效率最高。
所述HPLC-MS/MS对待测样品进行检测具体方法为:
液相色谱条件包括:
采用梯度洗脱,流动相A相:水(2mM乙酸铵),流动相B相:甲醇;
色谱柱为C18色谱柱;流动相流速为0.3~0.5ml/min(优选为0.4ml/min);柱温为25~40℃(优选为35℃);进样量0.5~2μL(优选为1μL);
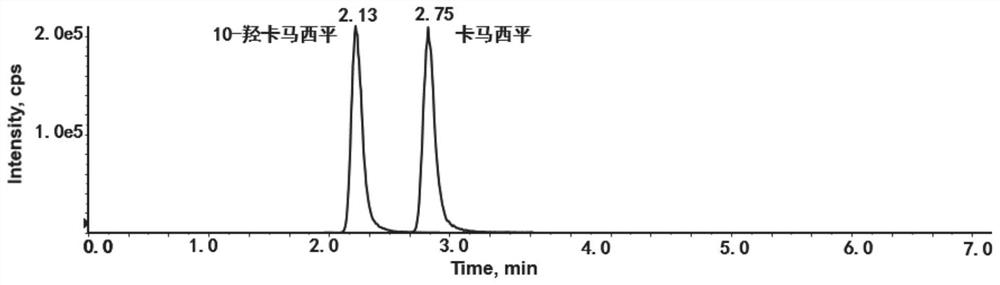
具体的,所述色谱柱为Kinetex Polor C18色谱柱,经研究发现,其可将卡马西平和10-羟卡马西平在较短的时间内分离,且峰形对称,方法可靠。在优化的高效液相色谱条件下,检测到卡马西平和10-羟卡马西平在保留时间为2.13和2.75分钟;总运行时间是4.5分钟。从而显著缩短检测时间。
梯度洗脱方式具体为:0-1.5min,流动相B 5-5%;1.5-1.9min,流动相B 5-70%;1.9-2.0min,流动相B 70-95%;2.0-3.5min,流动相B 95-95%;3.5-3.6min,流动相B 95-5%;3.6-4.5min,流动相B 5-5%。
质谱条件包括:
离子源:电喷雾(ESI);扫描方式:多反应监测(MRM);离子化方式:正离子;离子源电压:5000V;离子源温度:650℃;气帘气:15psi;雾化气:45psi;辅助气:55psi。
本发明的第二个方面,提供检测卡马西平和10-羟卡马西平的试剂盒,所述试剂盒包括,卡马西平和10-羟卡马西平标准品;
卡马西平-d10和10-羟卡马西平-d3内标标准品;
稀释液为乙腈或乙腈/甲醇混合溶液;
其中,所述乙腈/甲醇混合溶液的用量体积比为2~4:1~3,优选为3:2。
进一步的,所述试剂盒还包括空白血浆。
本发明的第三个方面,提供上述测定卡马西平和10-羟卡马西平的方法和/或检测试剂盒在药代动力学研究中的应用。
具体的,所述应用包括:基于上述检测方法对口服给药的人体药代动力学进行研究。
与现有技术相比,上述一个或多个技术方案存在如下有益技术效果:
上述技术方案提供了一种沉淀蛋白法结合HPLC-MS/MS测定抗癫痫药物卡马西平和10-羟卡马西平在人体血浆中含量的方法,从特异性、线性、灵敏度、准确度、精密度、基质效应、回收率和稳定性等方面对卡马西平和10-羟卡马西平在人血浆中的测定进行了充分的验证,最终应用于口服给药的人体药动学研究。上述技术方案首次提供了一种高效的卡马西平和10-羟卡马西平的蛋白沉淀提取方法,在人体药代动力学研究中显示出明显的优越性。
上述技术方案液质联用定量测定方法具有准确可靠、灵敏度高、专属性检测限和定量限更低等优点,对血浆中药物成分含量分析具有良好的实际应用之价值。
附图说明
构成本发明的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
图1为本发明实施例1中卡马西平、10-羟卡马西平、卡马西平-d10和10-羟卡马西平-d3的结构式及裂解方式(A:卡马西平,B:10-羟卡马西平,C:卡马西平-d10,D:10-羟卡马西平-d3);
图2为口服卡马西平、奥卡西平后的平均血浆浓度-时间曲线;
图3为本发明实施例1中建立的卡马西平和10-羟卡马西平色谱质谱图。
具体实施方式
应该指出,以下详细说明都是例示性的,旨在对本发明提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。
需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本发明的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单数形式也意图包括复数形式,此外,还应当理解的是,当在本说明书中使用术语“包含”和/或“包括”时,其指明存在特征、步骤、操作、器件、组件和/或它们的组合。应理解,本发明的保护范围不局限于下述特定的具体实施方案;还应当理解,本发明实施例中使用的术语是为了描述特定的具体实施方案,而不是为了限制本发明的保护范围。
如前所述,而当奥卡西平与卡马西平联用时,同时检测性质极为相近的卡马西平和10-羟卡马西平成为抗癫痫药物检测的新难题。
有鉴于此,本发明提供一种沉淀蛋白法结合HPLC-MS/MS测定抗癫痫药物卡马西平和10-羟卡马西平在人体血浆中含量的方法,对样品前处理方法和仪器检测条件等进行了优化,从特异性、线性、灵敏度、准确度、精密度、基质效应、回收率和稳定性等方面对卡马西平和10-羟卡马西平在人血浆中的测定进行了充分的验证,最终应用于口服给药的人体药动学研究。
本发明的一个具体实施方式中,所述定量测定方法,其步骤如下:
(1)分别用乙腈溶解精确称量的标准对照品卡马西平和10-羟卡马西平,得到卡马西平和10-羟卡马西平的储备液,卡马西平和10-羟卡马西平的最终浓度为1000μg/mL。将精确体积的0.10mL卡马西平和10-羟卡马西平标准溶液分别移入10mL容量瓶中,并用乙腈定容,得到卡马西平和10-羟卡马西平10μg/mL的工作液。用乙腈稀释,得到2、4、10、20、100、200、400和600ng/mL的工作液。同时,将精确重量的10.00mg卡马西平-d10和10-羟卡马西平-d3原料药转移到10mL容量瓶中,用一定体积乙腈溶解,制备1000μg/mL的内标储备液。卡马西平-d10和10-羟卡马西平-d3的工作液浓度为50ng/mL,稀释溶剂为乙腈(ACN):甲醇(MEOH)=3:2(V/V)。所有药物的储备液在4℃的避光容器中储存至少60天,无变化。
在0.1、0.2、0.25、0.5、1.0、2.5、5.0、10、20、25、30ng/mL的卡马西平和10-羟卡马西平血浆中药物浓度点中制备其校准标准。根据FDA关于选择质量控制点(QCs)的指南,为了进行准确度和精密度研究,QCs在4个浓度水平下制备为6个重复品,包括定量下限(LLOQ)、低(L:定义为LLOQ的三倍)、中(M:定义为中范围)和高(H:定义为高范围)。而对于其他试验(在志愿者样品分析期间),只使用了3种浓度水平(LQC、MQC和HQC)的样品。对于卡马西平和10-羟卡马西平,分别在0.1、0.25、2.5和25ng/mL下制备LLOQ、LQC、MQC和HQC。
将45μL人空白血浆置于2.0mL离心管中,加入精确体积5μL的卡马西平和10-羟卡马西平2-600ng/mL工作溶液,以获得卡马西平和10-羟卡马西平的0.1-30ng/mL血浆浓度。然后加入含50ng/mL卡马西平-d10和10-羟卡马西平-d3的150μL沉淀蛋白溶剂(ACN:MEOH=3:2)振荡10min提取分析物和同位素内标,在4℃下以14000rpm离心15min分离上部有机相和下部水相。将100μL上清液溶解于100μL水相中,并涡旋混合2分钟,得到对照品溶液;
(2)测定:将步骤(1)得到的对照品溶液进行HPLC-MS/MS分析,采用梯度洗脱,流动相A相:水(2mM乙酸铵),流动相B相:甲醇。
本发明的又一具体实施方式中,所述步骤(1)中将45μL人空白血浆置于2.0mL离心管中,加入精确体积5μL的卡马西平和10-羟卡马西平2-600ng/mL工作溶液,以获得卡马西平和10-羟卡马西平的0.1-30ng/mL血浆浓度。然后加入含50ng/mL卡马西平-d10和10-羟卡马西平-d3的100-200μL沉淀蛋白溶剂(ACN:MEOH=3:2,V/V)振荡10min提取分析物和同位素内标,在4℃下以13000-15000rpm离心10-20min分离上部有机相和下部水相。将100μL上清液溶解于100μL水相中,并涡旋混合2分钟,得到对照品溶液。
本发明为了获得满意的色谱行为和最大限度地提高分析物和同位素内标的电离响应,我们在流动相体系上进行了几次尝试。考虑到流动相pH范围的稳定性和消除色谱峰的分裂,在流动相中加入了2mM乙酸铵。以不同比例的甲醇-水和乙腈-水对分析物和同位素内标进行洗脱实验,发现甲醇比乙腈具有更低的背景噪声和更好的分辨率。艾杰尔飞诺美Kinetex Polor C18色谱柱对卡马西平和10-羟卡马西平有较好的保留作用,2mM乙酸铵对卡马西平和10-羟卡马西平的响应显著增强。由于10-羟卡马西平是在卡马西平结构上10位增加了羟基,10-羟卡马西平分子会在离子源部分发生断裂,导致产生卡马西平的定量母离子分子量。Kinetex Polor C18色谱柱可将卡马西平和10-羟卡马西平在较短的时间内分离(分离度大于1.5),且峰形对称,方法可靠。在优化的高效液相色谱条件下,检测到卡马西平和10-羟卡马西平在保留时间为2.13和2.75分钟。总运行时间是4.5分钟。
本发明利用HPLC-MS/MS分析和MS参数优化正离子模式,提高了MRM测量对ESI源的响应。对于卡马西平,MRM的碎片化转变为m/z 237.1到194.1(图1),对于卡马西平-d10,MRM的碎片化转变为m/z 247.1到204.1(图1),对于10-羟卡马西平为m/z 255.1到237.1(图1),对于10-羟卡马西平-d3为m/z 258.1到240.1(图1),每个转变的停留时间为300ms。每个化合物的碰撞能为22ev。分离电位保持在40V。适用于所有分析物的源参数为15psi的幕气、介质的碰撞气体(CAD)、温度为650℃、离子喷射电压为5000V、离子源气体为60psi。
在样品制备的优化方面,与乙酸乙酯液-液萃取法相比,蛋白质沉淀法具有精度高、回收率高、操作简单等优点。样品制备采用蛋白质沉淀法。卡马西平和10-羟卡马西平的定量限值可用于人血浆样品中药物动力学的定量分析。
最初,沉淀蛋白的溶剂是乙腈和甲醇,但这造成了卡马西平和10-羟卡马西平含量的部分损失,这可能是由于乙腈和甲醇不能有效地将分析物从蛋白质中解吸出来造成的。影响电荷态分布的因素包括溶剂pH值和药物溶解度。当使用乙腈:甲醇=3:2作为沉淀蛋白溶剂时,我们发现其沉淀效率最高,因此选择乙腈:甲醇=3:2作为沉淀蛋白溶剂。
本发明的又一具体实施方式中,上述步骤(2)中色谱柱为Kinetex Polor C18色谱柱(2.1mm×100mm,2.7μm,艾杰尔飞诺美公司);流动相流速为0.4ml/min;柱温为35℃;进样量1μL。
本发明试验了四个流速(0.3mL/min,0.4mL/min和0.5mL/min)对检测结果的影响。结果表明:流速为0.4mL/min时,分离效果最佳,各色谱峰保留时间适宜,分离度好,基线平稳,峰形对称,因此选择流速为0.4mL/min。
同时,本发明试验了四个不同柱温(如25℃,30℃,35℃和40℃)对质谱色谱检测结果的影响。结果显示柱温为35℃时,色谱峰保留时间适宜,基线平稳,各色谱峰分离度较好,峰形对称,因此选择柱温为35℃。
卡马西平、10-羟卡马西平、卡马西平-d10和10-羟卡马西平-d3的质谱参数见表1。
表1分析物和同位素内标的质谱参数
本发明同时对质谱条件进行优化,采用API5500型三重四级杆质谱仪的多反应离子检测模式(MRM)优化分析物和同位素内标的质谱条件,保证每一对离子对高响应的出峰,检测结果如表1所示,分析物和同位素内标均找到特异性的母离子、子离子用于定量分析。
在分析时间的选择上,本发明在选择色谱图谱的洗脱时间时记录了10min的色谱图。结果表明4.5min以后基本没有明显的色谱峰出现,同时为了照顾批次样品的差异性,保证所有批次样品的特征峰都能够被检出,因此选择4.5min作为分析时间。
本发明的又一具体实施方式中,步骤(3)中梯度洗脱方式为:0-1.5min,流动相B5-5%;1.5-1.9min,流动相B 5-70%;1.9-2.0min,流动相B 70-95%;2.0-3.5min,流动相B95-95%;3.5-3.6min,流动相B 95-5%;3.6-4.5min,流动相B 5-5%;
本发明的又一具体实施方式中,上述中质谱条件为:离子源:电喷雾(ESI);扫描方式:多反应监测(MRM);离子化方式:正离子;离子源电压:5000V;离子源温度:650℃;气帘气:15psi;雾化气:45psi;辅助气:55psi;
本发明的又一具体实施方式中,采用卡马西平-d10和10-羟卡马西平-d3作为同位素内标化合物。
本发明采用HPLC-MS/MS液质联用分析方法,实验过程中尝试选择离子检测(SIM)模式测定,结果各成分响应较低且基线高,基质影响较大,无法实现定量分析,然而通过多反应检测(MRM)法对母离子和特征碎片的子离子进行扫描时,发现离子峰的响应强度明显高于选择离子检测(SIM)模式,且基线低可实现定量分析。因此实验选用多反应检测(MRM)扫描模式用于定量卡马西平和10-羟卡马西平,用常规液相方法检测存在耗时长,分离较难,检测限高等缺点,不利于试验的进行。
以下通过实施例对本发明做进一步解释说明,但不构成对本发明的限制。应理解这些实施例仅用于说明本发明而不用于限制本发明的范围。
一种沉淀蛋白法结合HPLC-MS/MS测定抗癫痫药物卡马西平和10-羟卡马西平在人体血浆中含量的方法,包括如下步骤:
第一步:
对照品溶液制备:分别用乙腈溶解精确称量的标准对照品卡马西平和10-羟卡马西平,得到卡马西平和10-羟卡马西平的储备液,卡马西平和10-羟卡马西平的最终浓度为1000μg/mL。将精确体积的0.10mL卡马西平和10-羟卡马西平标准溶液分别移入10mL容量瓶中,并用乙腈定容,得到卡马西平和10-羟卡马西平10μg/mL的工作液。用乙腈稀释,得到2、4、10、20、100、200、400和600ng/mL的工作液。同时,将精确重量的10.00mg卡马西平-d10和10-羟卡马西平-d3原料药转移到10mL容量瓶中,用一定体积乙腈溶解,制备1000μg/mL的内标储备液。卡马西平-d10和10-羟卡马西平-d3的工作液浓度为50ng/mL,稀释溶剂为乙腈(ACN):甲醇(MEOH)=3:2。所有药物的储备液在4℃的避光容器中储存至少60天,无变化。
在0.1、0.2、0.25、0.5、1.0、2.5、5.0、10、20、25、30ng/mL的卡马西平和10-羟卡马西平血浆中药物浓度点中制备其校准标准。根据FDA关于选择质量控制点(QCs)的指南,为了进行准确度和精密度研究,QCs在4个浓度水平下制备为6个重复品,包括定量下限(LLOQ)、低(L:定义为LLOQ的三倍)、中(M:定义为中范围)和高(H:定义为高范围)。而对于其他试验(在志愿者样品分析期间),只使用了3种浓度水平(LQC、MQC和HQC)的样品。对于卡马西平和10-羟卡马西平,分别在0.1、0.25、2.5和25ng/mL下制备LLOQ、LQC、MQC和HQC。
将45μL人空白血浆置于2.0mL离心管中,加入精确体积5μL的卡马西平和10-羟卡马西平2-600ng/mL工作溶液,以获得卡马西平和10-羟卡马西平的0.1-30ng/mL血浆浓度。然后加入含50ng/mL卡马西平-d10和10-羟卡马西平-d3的150μL沉淀蛋白溶剂(ACN:MEOH=3:2,V/V)振荡10min提取分析物和同位素内标,在4℃下以14000rpm离心15min分离上部有机相和下部水相。将100μL上清液溶解于100μL水相中,并涡旋混合2分钟,得到对照品溶液;
第二步:
测定:将步骤(1)得到的对照品溶液进行HPLC-MS/MS分析,采用梯度洗脱,流动相A相:水(2mM乙酸铵),流动相B相:甲醇。
在本实施例中,色谱柱为Kinetex Polor C18色谱柱(2.1mm×100mm,2.7μm,艾杰尔飞诺美公司);流动相流速为0.4ml/min;柱温为35℃;进样量1μL。各有效成分质谱参数见表1。梯度洗脱方式为:0-1.5min,流动相B 5-5%;1.5-1.9min,流动相B 5-70%;1.9-2.0min,流动相B 70-95%;2.0-3.5min,流动相B 95-95%;3.5-3.6min,流动相B 95-5%;3.6-4.5min,流动相B 5-5%。图3为本发明建立的卡马西平和10-羟卡马西平色谱质谱图。
质谱条件为:离子源:电喷雾(ESI);扫描方式:多反应监测(MRM);离子化方式:正离子;离子源电压:5000V;离子源温度:650℃;气帘气:15psi;雾化气:45psi;辅助气:55psi;
第三步:
对建立的高效液相串联质谱的方法可行性进行考察的方法包括对专属性、定量限、精密度、准确度、稳定性、基质效应及提取回收率。
专属性:通过比较六个人的空白血浆样品、在最后给药后0.5小时从其中一名受试者获得的临床血浆样品、以30ng/mL的剂量加入卡马西平和10-羟卡马西平的血浆样品和以0.1ng/mL的剂量加入卡马西平和10-羟卡马西平的血浆样品的色谱图来评估特异性和内源性干扰。通过比较50%乙腈和添加卡马西平和10-羟卡马西平的三蒸馏水(0.1ng/mL)的LLOQ和同位素内标(50ng/mL)的色谱图来评估特异性和外源性干扰。制备并分析所有空白血浆样品,以确保没有干扰峰。在所建立的色谱条件下,血浆中没有内源性干扰,表明该方法的选择性是可以接受的;
定量限:采用1/X
精密度:在同一天对四种浓度(0.1、0.25、2.5和25ng/mL)的六个重复的LLOQ和QC样品进行分析,以评估日内精密度和准确度。通过连续三天分析LLOQ和QC样本来评估日间精密度和准确度。方法的精密度和准确度分别用相对标准偏差(RSD)和相对误差(RE)表示。RSD和RE均不得超过15%。然而,在LLOQ,RE和RSD<±20%是可以接受的。LLOQ和QC样品中卡马西平和10-羟卡马西平的精密度和准确度结果见表2。卡马西平和10-羟卡马西平各样品水平的精密度(RSD)均小于9.99%。卡马西平和10-羟卡马西平各样品水平的准确度在1.48%到8.31%之间,测定值均在可接受范围内。
表2人血浆中测定卡马西平和10-羟卡马西平含量方法的精密度和准确度
基质效应和提取回收率:提取回收率是通过比较在6个不同批次血浆中制备的三个水平的QC样品中提取的分析物与IS比的绝对峰面积与空白血浆、高溶血性血浆和高脂肪血浆提取后用相同浓度的分析物纯溶液进行强化LQC、MQC、HQC。通过比较六批样品在LQC、MQC、HQC水平下强化的空白血浆提取物与相同浓度水平分析物强化的空白水提取物中分析物绝对峰面积与IS比值的比较,来评估基质效应。在人空白血浆中,卡马西平和10-羟卡马西平内标物均匀化的平均基质效应为101.2-106.0%,而高溶血性的平均基质效应为99.9-101.4%。在高脂血浆中,卡马西平和10-羟卡马西平的基质效应为95.5-101.1%。如表3所示,所有相对标准偏差值在0.75%到9.17%之间,这表明血浆基质的影响对于分析来说是可以忽略的。卡马西平和10-羟卡马西平内标物均匀化后的平均提取回收率为96.7-103.8%,不同浓度下卡马西平和10-羟卡马西平的提取回收率结果准确、重现性好。
表3人血浆中测定卡马西平和10-羟卡马西平含量方法的提取回收率和基质效应(n=6)。
稳定性试验:对三种不同浓度的QC样品在不同条件下的稳定性进行了分析:(1)连续三次冻融循环(从-20℃到23℃);(2)室温(23℃)制备前3h;(3)冰箱温度(4℃)制备后20小时,室温(23℃)制备后6小时;(4)自动进样器10℃制备后24小时;(5)冰箱温度(-20℃)制备前3、8、31天。通过比较储存的QC样品和新制备的样品的平均浓度来评估溶液的稳定性。样品被认为是稳定的,与标称浓度的偏差在±15.0%以内。所有稳定性试验样品在6个重复中进行分析,并根据新制备的样品确定偏差。卡马西平和10-羟卡马西平在三次冻融和室温下放置至少3h后,卡马西平和10-羟卡马西平的CV%(7.35%)的响应无显著差异(<15%),表明卡马西平和10-羟卡马西平在此条件下是稳定的。血浆样品在至少三个冷冻/解冻循环中的CV值(11.92%)稳定。处理后的样品在自动进样器中稳定达24小时,在室温托盘中稳定达3小时,CV%值分别至少为6.88%和7.35%。血浆样品在-20℃下稳定至少4周,无明显损失(<8.43%)。结果见表4和表5。
表4卡马西平和10-羟卡马西平的样品稳定性(n=6,以Mean±R.E.%表示)
表5卡马西平和10-羟卡马西平溶液稳定性(n=6)
一种沉淀蛋白法结合HPLC-MS/MS测定抗癫痫药物卡马西平和10-羟卡马西平在人体血浆中含量的方法及其应用,包括如下步骤:
第一步:
对12名健康男性受试者进行了药代动力学研究。伦理委员会批准了方案,志愿者得到了知情的书面同意。给药前禁食12小时,给药后禁食3小时。口服给药卡马西平和奥卡西平后,在给药前和0时从颈静脉抽取2mL的血样。后分别于0.083,0.167,0.333,0.5,0.75,1,1.25,1.75,2,2.5、3、4、5、6、8、12和24小时取血。实验期间,可以自由饮水。随后在14000×g转速下离心10分钟制备血浆,并在-80℃下立即冷冻。
将50μL人空白血浆置于2.0mL离心管中,加入含50ng/mL同位素内标的150μL沉淀蛋白溶剂(ACN:MEOH=3:2,V/V)振荡10min提取分析物和同位素内标,在4℃下以14000rpm离心15min分离上部有机相和下部水相。将100μL上清液溶解于100μL水相中,并涡旋混合2分钟,得到供试品溶液;
第二步:
测定:将步骤(1)得到的供试品溶液进行HPLC-MS/MS分析,采用梯度洗脱,流动相A相:水(2mM乙酸铵),流动相B相:甲醇。
药代动力学分析采用DAS2非房室模型软件程序(中国数学药理学专业委员会,中国上海)计算AUC、C
口服卡马西平和奥卡西平后的平均血浆浓度-时间曲线如图2所示。药代动力学参数见表6。
表6口服卡马西平和奥卡西平后的非房室药代动力学参数(平均值±标准差,n=12)
应注意的是,以上实例仅用于说明本发明的技术方案而非对其进行限制。尽管参照所给出的实例对本发明进行了详细说明,但是本领域的普通技术人员可根据需要对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的精神和范围。
- 一种测定卡马西平和10-羟卡马西平的方法、试剂盒及其应用
- 一种定量测定人血浆中卡马西平的方法及应用