一种何首乌中二蒽酮类化合物的检测方法
文献发布时间:2023-06-19 11:26:00
技术领域
本发明涉及化学分析及检测技术领域,尤其涉及一种何首乌中二蒽酮类化合物的检测方法。
背景技术
何首乌为生熟异治的典型品种之一,分为生何首乌和制何首乌。生何首乌为蓼科植物何首乌属(Fallopia)何首乌(Polygonum multiflorum Thunb.)的干燥块根,具有解毒、消痈、截疟、润肠通便等功效,常用于治疗疮痈,风疹瘙痒,久疟体虚及肠燥便秘。制何首乌为何首乌的炮制加工品,具有补肝肾、益精血、乌须发、强筋骨、化浊降脂等功效,常用于治疗血虚微黄、眩晕耳鸣、须发早白、腰膝酸软、肢体麻木及高血脂症等。
《本草汇言》记载何首乌:“生用气寒,性敛,有毒;制熟,气温无毒”。何首乌中化学成分种类较多,迄今已发现150多种化合物,主要为二苯乙烯类、蒽醌类、二蒽酮类、多酚类及萘类等化学成分。近年来,何首乌在临床上的肝毒性等不良反应报道,不断增多,一定程度上影响了何首乌的应用。研究发现,何首乌中二蒽酮类成分,具有一定的肝毒性,尤其反式-大黄素二蒽酮(trans-emodin dianthrones)和顺式-大黄素二蒽酮(cis-emodindianthrones),在斑马鱼的胚胎发育和肝毒性评价模型中,均表现一定的毒性作用,提示该类成分可能为何首乌中潜在的肝毒性成分。
所以,有必要增加何首乌中二蒽酮类成分的限量标准。目前,有关采用超高效液相色谱-串联四级杆质谱联用技术测定何首乌中二蒽酮类成分的方法未见报道。
发明内容
有鉴于此,本发明的目的在于提供一种何首乌中二蒽酮类化合物的检测方法。本发明利用超高效液相色谱-串联四级杆质谱联法对何首乌中的二蒽酮类化合物进行检测,具有灵敏度高的优势。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种何首乌中二蒽酮类化合物的检测方法,包括以下步骤:
将何首乌、内标物和提取剂混合,经超声,得到待测液;所述提取剂为体积浓度为70%的乙醇溶液;
利用超高效液相色谱-串联四级杆质谱联法对所述待测液进行检测,得到何首乌中各二蒽酮类化合物峰面积比值;
基于各二蒽酮类化合物浓度-峰面积比值的标准曲线,获得何首乌中二蒽酮类化合物的含量;
所述二蒽酮类化合物包括PolygonumnolideC4、PolygonumnolideC3、PolygonumnolideC1、PolygonumnolideC2、反式大黄素二蒽酮和顺式大黄素二蒽酮;
所述超高效液相色谱-串联四级杆质谱联法包括色谱条件和质谱条件;
所述色谱条件包括以下参数:
色谱柱:AgilentZORBAX SB-C
柱温:30±5℃;
流量:0.25mL/min;
流动相A:乙腈;
流动相B:质量浓度为0.1%的甲酸水溶液;
梯度洗脱程序:
0~8min:流动相A为37%;
8~10min:流动相A由37%增加至60%;
10~12min:流动相A由60%增加至78%;
12~20min:流动相A由78%增加至90%;
20~22min:流动相A由90%降低至37%;
22~30min:流动相A维持在37%;
进样量:2.0μL;
所述质谱条件包括以下参数:
模式:负离子模式;
扫描方式:多反应检测模式;
喷射电压:4.0kV;
离子源温度:300℃;
雾化器的压力:30psi;
流速:10L/min;
气氛:高纯氮气。
优选地,所述内标物为二蒽酮基。
优选地,所述待测液中内标物的浓度为5~10μg/mL。
优选地,所述何首乌和提取剂的用量比为1g:(40~50)mL。
优选地,所述何首乌粒径≤50目。
优选地,所述超声的功率为250~1000W,频率为30~50kHz,时间为20~40min。
本发明提供了一种何首乌中二蒽酮类化合物的检测方法,包括以下步骤:将何首乌、内标物和提取剂混合,经超声,得到待测液;所述提取剂为体积浓度为70%的乙醇溶液;利用超高效液相色谱-串联四级杆质谱联法对所述待测液进行检测,得到何首乌中各二蒽酮类化合物峰面积比值;基于各二蒽酮类化合物浓度-峰面积比值的标准曲线,获得何首乌中二蒽酮类化合物的含量;所述二蒽酮类化合物包括PolygonumnolideC4、PolygonumnolideC3、PolygonumnolideC1、PolygonumnolideC2、反式大黄素二蒽酮和顺式大黄素二蒽酮;所述超高效液相色谱-串联四级杆质谱联法包括色谱条件和质谱条件;所述色谱条件包括以下参数:色谱柱:AgilentZORBAX SB-C
本发明以体积浓度为70%的乙醇对何首乌中的二蒽酮类化合物进行提取,提取效果率高;本发明采用内标法对何首乌中的二蒽酮类化合物进行检测,提高了检测灵敏度。另外,选择乙腈和质量浓度为0.1%的甲酸水溶液作为流动相,使何首乌中的二蒽酮类化合物具有很好的分离度,提高了检测灵敏度。本发明采用电喷雾阴离子串联质谱多反应检测(MRM)模式,快速在线优化包括碎裂电压以及碰撞能在内的质谱参数,使之形成高强度的靶向离子对,通过此系列特征性的靶向离子对,从而达到对何首乌药材中的二蒽酮类化合物的快速定性和定量分析。
附图说明
图1为实施例2所得何首乌待测液的总离子流图;
图2~4为实施例2所得何首乌待测液的选择离子流图;
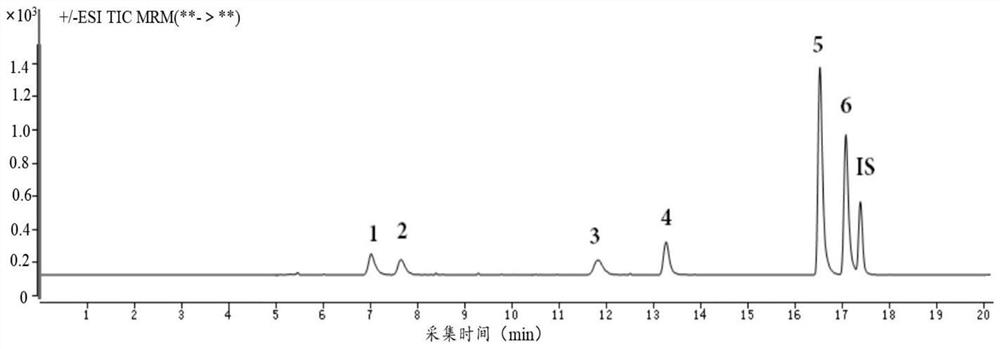
图5为对比例2的总离子流色谱图;
图6为对比例3的总离子流色谱图;
其中,1为PolygonumnolideC4、2为PolygonumnolideC3、3为PolygonumnolideC1、4为PolygonumnolideC2、5为反式大黄素二蒽酮,6为顺式大黄素二蒽酮,IS为二蒽酮基。
具体实施方式
本发明提供了一种何首乌中二蒽酮类化合物的检测方法,包括以下步骤:
将何首乌、内标物和提取剂混合,经超声,得到待测液;所述提取剂为体积浓度为70%的乙醇溶液;
利用超高效液相色谱-串联四级杆质谱联法对所述待测液进行检测,得到何首乌中各二蒽酮类化合物峰面积比值;
基于各二蒽酮类化合物浓度-峰面积比值的标准曲线,获得何首乌中二蒽酮类化合物的含量。
本发明将何首乌、内标物和提取剂混合,经超声,得到待测液。
在本发明中,所述二蒽酮类化合物包括具有式I所示结构的PolygonumnolideC4、具有式II所示结构的PolygonumnolideC3、具有式III所示结构的PolygonumnolideC1、具有式IV所示结构的PolygonumnolideC2、具有式V所示结构的反式大黄素二蒽酮二蒽酮和具有式VI所示结构的顺式二蒽酮二蒽酮。
在本发明中,所述何首乌的粒径优选≤50目。在本发明中,所述内标物优选为二蒽酮基,具有式VII所示结构。
在本发明中,所述内标物以内标物溶液的形式加入,所述内标物溶液的溶剂优选为体积比2:1的DMSO-甲醇溶剂;本发明对所述内标物溶液的浓度和加入量不做具体限定,只要能够使待测液中内标物的浓度为5~10μg/mL,进一步优选为6.3μg/mL。
在本发明中,所述提取剂为体积浓度为70%的乙醇水溶液。在本发明中,所述何首乌和提取剂的用量比优选为1g:(40~50)mL,具体优选为1:50mL。
在本发明中,所述超声的功率优选为250~1000W,进一步优选为500W;频率优选为30~50kHz,进一步优选为40kHz;时间优选为20~40min,进一步优选为30min。
超声结束后,本发明优选还包括将所得超声体系过滤,所述过滤用滤膜的孔径优选为0.22μm。
本发明以体积浓度为70%的乙醇溶液作为提取剂,对何首乌中二蒽酮类化合物的提取效率高;另外添加内标物二蒽酮基,提高了检测灵敏度。
得到待测液后,本发明利用超高效液相色谱-串联四级杆质谱联法对所述待测液进行检测,得到何首乌中各二蒽酮类化合物峰面积比值。
在本发明中,所述超高效液相色谱-串联四级杆质谱联法包括色谱条件和质谱条件;
所述色谱条件包括以下参数:
色谱柱:Agilent ZORBAX SB-C
柱温:30±5℃;
流量:0.25mL/min;
流动相A:乙腈;
流动相B:质量浓度为0.1%的甲酸水溶液;
梯度洗脱程序:
0~8min:流动相A为37%;
8~10min:流动相A由37%增加至60%;
10~12min:流动相A由60%增加至78%;
12~20min:流动相A由78%增加至90%;
20~22min:流动相A由90%降低至37%;
22~30min:流动相A维持在37%;
进样量:2.0μL;
所述质谱条件包括以下参数:
模式:负离子模式;
扫描方式:多反应检测模式;
喷射电压:4.0kV;
离子源温度:300℃;
雾化器的压力:30psi;
流速:10L/min;
气氛:高纯氮气。
在本发明中,所述PolygonumnolideC4、PolygonumnolideC3、PolygonumnolideC1、PolygonumnolideC2、反式大黄素二蒽酮、顺式大黄素二蒽酮和二蒽酮基的MRM参数如表1所示。
表1二蒽酮类化合物和二蒽酮基的MRM参数
注:表1中,1为PolygonumnolideC4、2为PolygonumnolideC3、3为PolygonumnolideC1、4为PolygonumnolideC2、5为反式大黄素二蒽酮,6为顺式大黄素二蒽酮,7为二蒽酮基。
在本发明中,得到的何首乌中各二蒽酮类化合物峰面积比值优选为待测物与内标物的峰面积比值,具体优选为PolygonumnolideC4和内标物的峰面积比值、PolygonumnolideC3和内标物的峰面积比值、PolygonumnolideC1和内标物的峰面积比值、PolygonumnolideC2和内标物的峰面积比值、反式大黄素二蒽酮和内标物的峰面积比值、顺式大黄素二蒽酮和内标物的峰面积比值。
得到何首乌中各二蒽酮类化合物峰面积比值后,本发明基于各二蒽酮类化合物浓度-峰面积比值的标准曲线,获得何首乌中二蒽酮类化合物的含量。
在本发明中,所述各二蒽酮类化合物浓度-峰面积比值的标准曲线分别为:PolygonumnolideC4浓度-峰面积比值标准曲线、PolygonumnolideC3浓度-峰面积比值标准曲线、PolygonumnolideC1浓度-峰面积比值标准曲线、PolygonumnolideC2浓度-峰面积比值标准曲线、反式大黄素二蒽酮浓度-峰面积比值标准曲线和顺式大黄素二蒽酮浓度-峰面积比值标准曲线。
本发明对所述各二蒽酮类化合物浓度-峰面积比值的标准曲线的获取方式不做具体限定,采用本领域技术人员熟知的内标法标准曲线的获取方法进行绘制即可。
下面结合实施例对本发明提供的何首乌中二蒽酮类化合物的检测方法进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
以下实施例利用超高效液相色谱-串联四级杆质谱联法对所述待测液进行检测的参数均为:
所述色谱条件包括以下参数:
色谱柱:Agilent ZORBAX SB-C
柱温:30±5℃;
流量:0.25mL/min;
流动相A:乙腈;
流动相B:质量浓度为0.1%的甲酸水溶液;
梯度洗脱程序:
0~8min:流动相A为37%;
8~10min:流动相A由37%增加至60%;
10~12min:流动相A由60%增加至78%;
12~20min:流动相A由78%增加至90%;
20~22min:流动相A由90%降低至37%;
22~30min:流动相A维持在37%;
进样量:2.0μL;
所述质谱条件包括以下参数:
模式:负离子模式;
扫描方式:多反应检测模式;
喷射电压:4.0kV;
离子源温度:300℃;
雾化器的压力:30psi;
流速:10L/min;
气氛:高纯氮气。
实施例1
对照品溶液的制备:
分别精密称取对照品PolygonumnolideC4、PolygonumnolideC3、PolygonumnolideC1、PolygonumnolideC2、反式大黄素二蒽酮和顺式大黄素二蒽酮,用体积浓度为70%的乙醇溶解,配制成PolygonumnolideC4浓度为0.210μg/mL、PolygonumnolideC3浓度为0.214μg/mL、PolygonumnolideC1浓度为0.283μg/mL、PolygonumnolideC2浓度为0.280μg/mL、反式大黄素二蒽酮浓度为0.318μg/mL和顺式大黄素二蒽酮浓度为0.280μg/mL的混合标准贮备液。
精密称取内标物二蒽酮基25.11mg,加DMSO/甲醇(其中DMSO和甲醇的体积比为2:1)溶解,得到浓度为100.44μg/mL的内标物溶液。
将混合标准贮备液中各物质的浓度分别稀释至0.6倍、0.4倍、0.2倍、0.1倍、0.08倍、0.04倍、0.02倍和0.01倍,得到系列混合标准液;在检测前分别在1.5mL的系列标准混液中,加入0.1mL的内标物溶液进行检测;以混合标准液的质量浓度作为横坐标(X),以各对照品和内标物二蒽酮基的峰面积比作为纵坐标(Y),分别得到6个对照片的线性回归方法,结果见表2。
按照检测信号高度为与基线噪音高度的比值(信噪比)为3:1,确定检出限(LOD);结果如表2所示。
按照检测信号高度为与基线噪音高度的比值(信噪比)为10:1,确定定量限(LOQ),结果如表2所示。
表2何首乌中二蒽酮类化合物的回归方程、相关系数、线性范围、定量限和检出限汇总结果
注:表2中,1为PolygonumnolideC4、2为PolygonumnolideC3、3为PolygonumnolideC1、4为PolygonumnolideC2、5为反式大黄素二蒽酮,6为顺式大黄素二蒽酮。
由表2可以看出:何首乌中6个二蒽酮类化合物的含量在线性范围内具有良好的相关系数(R
实施例2
精密称定何首乌药材粉末(过三号筛)约1.0g,置具塞锥形瓶中,精密加50mL体积浓度为70%的乙醇溶液,称定重量,超声处理(功率500W,频率40kHz)30分钟,取出,放冷,再称定重量,用体积浓度为70%的乙醇溶液补足减失的重量,摇匀,滤过,过0.22μm的滤膜,取续滤液即得。在1.5mL滤液中加入0.1mL内标物溶液进行检测,所得何首乌待测液的总离子流图如图1所示;图2~4为所得何首乌待测液的选择离子流图;图1~4中,1为PolygonumnolideC4、2为PolygonumnolideC3、3为PolygonumnolideC1、4为PolygonumnolideC2、5为反式大黄素二蒽酮二蒽酮,6为顺式大黄素二蒽酮二蒽酮,IS为二蒽酮基。从图1~图4可以看出:本发明提供的检测方法能够同时测定六种二蒽酮类化合物的含量。
实施例3
3.1精密度试验
按照实施例2公开的方法制备何首乌待测液,利用超高效液相色谱-串联四级杆质谱联法对何首乌待测液分别在24h之内连续充分进样6次,连续72h每天重复进样3次,通过其对应的峰面积比值和保留时间的相对标准偏差(RSD)进行计算,结果见表3。
表3何首乌中二蒽酮类化合物的精密度试验结果
注:表3中,1为PolygonumnolideC4、2为PolygonumnolideC3、3为PolygonumnolideC1、4为PolygonumnolideC2、5为反式大黄素二蒽酮,6为顺式大黄素二蒽酮。
从表3可以看出:何首乌待测液的日内和日间的相对峰面积比值的相对标准偏差(RSD),均小于5.0%,表明本发明提供的检测方法具有良好的精密度。
3.2重复性试验
取同一批次何首乌药材粉末1份,按照实施例2的方法制备何首乌待测液,利用超高效液相色谱-串联四级杆质谱联法,连续进样6次,进行检测,计算6个二蒽酮类化合物的峰面积与内标物的峰面积比值,结果如表4所示。
表4何首乌中二蒽酮类化合物的重复性试验结果
注:表4中,1为PolygonumnolideC4、2为PolygonumnolideC3、3为PolygonumnolideC1、4为PolygonumnolideC2、5为反式大黄素二蒽酮,6为顺式大黄素二蒽酮。
从表4可以看出:何首乌待测液的相对峰面积比值的相对标准偏差(RSD)范围为1.73~3.24%,均不超过5.0%,表明该方法具有良好的重复性。
3.3重现性试验
取6个不同批次何首乌药材粉末各一份,按照实施例2的方法制备何首乌待测液,利用超高效液相色谱-串联四级杆质谱联法进行检测,计算6个二蒽酮类化合物的含量,结果如表5所示。
表5何首乌中二蒽酮类化合物的重现性试验结果
注:表5中,1为PolygonumnolideC4、2为PolygonumnolideC3、3为PolygonumnolideC1、4为PolygonumnolideC2、5为反式大黄素二蒽酮,6为顺式大黄素二蒽酮。
由表5可知,何首乌待测液的峰面积比值的相对标准偏差(RSD)范围为1.23~3.30%,均不超过4.0%,说明本发明提供的检测方法具有良好的重复性。
3.4稳定性试验
按照实施例2中的方法制备何首乌待测液,以24h为一个周期,分别在0、2、4、8、12和24h进样一次,计算6个二蒽酮类化合物的峰面积与内标物的峰面积比值,并对其峰面积比值的相对标准偏差(RSD)进行计算,结果见表6。
表6何首乌中二蒽酮类化合物的稳定性试验结果
注:表6中,1为PolygonumnolideC4、2为PolygonumnolideC3、3为PolygonumnolideC1、4为PolygonumnolideC2、5为反式大黄素二蒽酮,6为顺式大黄素二蒽酮。
从表6可以看出,何首乌待测液的相对峰面积比值的相对标准偏差(RSD)范围为3.41~3.95%,均不超过4.0%,说明本发明提供的检测方法具有良好的稳定性。
3.5加样回收率试验
取已知6个二蒽酮类化合物含量的何首乌粉末6份,精密称取何首乌粉末0.5g,加入6个二蒽酮类化合物混合标准贮备液,按何首乌待测液的制备方法进行制备,利用超高效液相色谱-串联四级杆质谱联法进行检测,计算回收率,结果如表7所示。
表7何首乌中二蒽酮类化合物的加样回收率试验结果
注:表7中,1为PolygonumnolideC4、2为PolygonumnolideC3、3为PolygonumnolideC1、4为PolygonumnolideC2、5为反式大黄素二蒽酮,6为顺式大黄素二蒽酮。
从表7可以看出:6个二蒽酮类化合物的加样回收率在104.38~150.4%,表明该方法具有较好的回收率。
加收回收率=(M
实施例4
称取20批不同批次的何首乌1.0g,精密称定;
按下述方法制备待测液:取何首乌药材粉末(过三号筛)约1.0g,置具塞锥形瓶中,精密加体积浓度为70%的乙醇溶液50mL,称定重量,超声处理(功率100W,频率40kHz)30分钟,取出,放冷,再称定重量,用70%乙醇补足减失的重量,摇匀,滤过,过0.22μm的滤膜,取续滤液即得待测液。
在1.5mL待测液中加入0.1mL内标物溶液,得到何首乌待测液;
采用上述条件下进样检测,记录色谱图,检测结果见表8。
表8 20批何首乌样品中6个二蒽酮类化合物含量(μg/g)
注:表8中,1为PolygonumnolideC4、2为PolygonumnolideC3、3为PolygonumnolideC1、4为PolygonumnolideC2、5为反式大黄素二蒽酮,6为顺式大黄素二蒽酮。
从表8可以看出,在不同产地的20批何首乌药材中,PolygonumnolideC4(1),PolygonumnolideC3(2),PolygonumnolideC1(3),PolygonumnolideC2(4),反式大黄素二蒽酮(5)和顺式大黄素二蒽酮(6)总体含量都很低;化合物5和6的含量,明显高于化合物1~4;6种二蒽酮类化合物在20批药材中总含量范围为1.98μg/g~143.17μg/g。
对比例1
精密称定何首乌药材粉末(过三号筛)约1.0g,置具塞锥形瓶中,精密加50mL体积浓度分别为水、30%乙醇、50%乙醇、70%乙醇和95%乙醇溶液,称定重量,超声处理(功率500W,频率40kHz)30分钟,取出,放冷,再称定重量,用体积浓度为所提溶液补足减失的重量,摇匀,滤过,过0.22μm的滤膜,取续滤液即得。在1.5mL滤液中加入0.1mL内标物溶液,得到待测液进行测定。结果如表9所示。
表9不同提取溶剂的优化结果
从表9可以看出:本发明采用的体积浓度为70%的乙醇水溶液超声提取法所得6种二蒽酮类化合物的相对峰面积较高,提取效率较好。
对比例2
精密称定何首乌药材粉末(过三号筛)约1.0g,置具塞锥形瓶中,精密加25mL、50mL、100mL和150mL体积浓度为70%的乙醇溶液,称定重量,超声处理(功率500W,频率40kHz)30分钟,取出,放冷,再称定重量,用体积浓度为70%的乙醇溶液补足减失的重量,摇匀,滤过,过0.22μm的滤膜,取续滤液即得。在1.5mL滤液中加入0.1mL内标物溶液,得到待测液进行测定。
结果如表10和图5所示。
表10不同提取溶剂体积的优化结果
从表10和图5可以看出:本发明采用的50mL体积浓度为70%的乙醇水溶液超声提取法所得6种二蒽酮类化合物的相对峰面积较高,提取效率较好。
对比例3
精密称定何首乌药材粉末(过三号筛)约1.0g,置具塞锥形瓶中,精密加50ml体积浓度为70%的乙醇溶液,称定重量,超声处理(功率500W,频率40kHz)15、30和45分钟,取出,放冷,再称定重量,用体积浓度为70%的乙醇溶液补足减失的重量,摇匀,滤过,过0.22μm的滤膜,取续滤液即得。在1.5mL滤液中加入0.1mL内标物溶液,得到待测液进行测定。
结果如表11和图6所示。
表11超声提取的不同时间优化结果
从表11和图6可以看出:本发明采用的50ml体积浓度为70%的乙醇水溶液超声提取30分钟法所得6种二蒽酮类化合物的相对峰面积较高,提取效率较好。
由以上实施例可知,本发明运用超高效液相色谱-串联四级杆质谱联用法,采用电喷雾阴离子串联质谱多反应检测(MRM)模式,快速在线优化包括碎裂电压以及碰撞能在内的质谱参数,使之形成高强度的靶向离子对,通过此系列特征性的靶向离子对,从而达到对何首乌药材中的二蒽酮类化合物的快速定性和定量分析;本发明的检测方法操作简单,具有高灵敏度和精密度、重现性好、稳定性高、回收率好,可实现同时测定何首乌中所含的二蒽酮类化合物,弥补现有何首乌药材质量控制技术不够完善和科学的不足,能准确、全面地控制制何首乌药材的质量。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 一种何首乌中二蒽酮类化合物的检测方法
- 一种检测印刷包装材料中二苯甲酮和异丙基硫杂蒽酮类化合物的合相色谱分析方法