具有抗肿瘤效果的咪唑并恶唑衍生物及包括其的药物组合物
文献发布时间:2023-06-19 11:54:11
技术领域
本公开涉及一种咪唑并恶唑衍生物(Imidazo[2,1-b]oxazole)或其药学上可接受的盐、其制备方法以及包括其作为有效成分并用于预防或治疗肿瘤的药物组合物。
背景技术
MAPK信号传导路径(EGFR>Ras>Raf>Mek>Erk)是关于细胞增殖的信号传导体系,其自1990年代被发现以后,在许多研究中已被观察为癌症发生的主要位置。
人体中存在三种已知的Raf激酶酵素(马雷和马歇尔癌症调查(Marais andMarshall Cancer Surv).27:101-125,1996),包括A-RAF、B-RAF以及C-RAF。随着发现B-RAF的突变种在人类肿瘤中占据高频率,因此,RAF蛋白被认为是恶性肿瘤的重要起源和促进因子(戴维斯,H.等人,自然417:949-954(2002))。
在B-RAF的情况下,发现其大约7%的突变与肿瘤有关,并分別显示了黑色素瘤(50%至70%)、卵巢癌(35%)、甲状腺癌(50%)和直肠癌(10%)的频率(图为森(Tuveson),等人,癌细胞.4:95-98(2003);及邢(Xing),内分泌相关癌症:12:245-262(2005)。这些突变中约有90%是激酶结构域的缬氨酸残基已点突变为谷氨酸的V600E,所述V600E成为抗癌剂开发的重要靶点。V600E BRAF具有超过500倍的磷酸化能,并产生MEK和ERK的过度活性化,导致肿瘤细胞的异常生长。至今确认的B-RAF突变种约为40个(主要发生在位于激酶活性结构域的活性片段部位和富含甘氨酸的G环中),并且除V600E之外的其他突变种的发生频率非常低。直肠癌中约10%的B-RAF突变种发生在激酶结构域的G环中(拉贾戈帕兰(Rajagopalan)等人,自然2002 418,934)。
在最近的研究中,报告了在人体的黑色素瘤细胞中利用小干扰RNA(siRNA)抑制突变的B-RAF的情况下,MEK和ERK都被抑制,肿瘤细胞的生长停止,最终促进了细胞的死亡(沙玛(Sharma),等人,癌症研究.65:2412-2421(2005);及威尔布洛克(Wellbrock)等人,癌症研究.64:2338-2342(2004))。此外,在使用短发夹RNA(shRNA)的B-RAF突变的异种移植模型的实验中,也显示出抑制B-RAF来抑制了肿瘤并且可逆地调节(赫夫利希(Hoeflich)等人,癌症研究.66:999-1006(2006))。综上所述,可以确认B-RAF细胞内的信号传导系与肿瘤的发生密切相关,并且B-RAF是抗癌剂的重要靶点。
发明内容
技术问题
根据本公开的一个方面,提供了咪唑并恶唑衍生化合物、溶剂化物、立体异构体或其药学上可接受的盐。
根据本公开的另一个方面,提供了包括所述化合物、溶剂化物、立体异构体或其药学上可接受的盐作为有效成分的用于预防或治疗肿瘤的药物组合物。
另一方面提供用于制备用于预防或治疗肿瘤的药物(medicament)的所述药学组合物的用途。
根据本公开的另一个方面,提供了包括将所述药物组合物施用于患上肿瘤或有患上肿瘤风险的个体的预防或治疗肿瘤的方法。
技术方案
根据本公开的一个方面,由以下化学式P表示的化合物、溶剂化物、立体异构体或药学上可接受的盐,
在所述化学式P中,
X为O或S,
A为
R
Y可为氢、羟基、C
B是
n=1或2,
R
R
所述取代基可选自卤素、C
在一个实施例中,R
Y可为氢、羟基、C
R
R
所述一个以上的取代基可以为选自卤素﹑C
在另一实施例中,所述A的苯基可具有多个取代基。例如,所述A的苯基可以具有1、2、3、4,5个的取代基。
所述“卤素”可为氟(F)、Cl、Br或I。
所述“烷氧基”可为甲氧基、乙氧基、n-丙氧基、异丙氧基、n-丁氧基、异丁氧基或t-丁氧基。
所述“烷基”是指具有特定碳原子数的直链或支链的脂肪族烃基基团,可为甲基、乙基、n-丙基、异丙基、丁基、异丁基或t-丁基。
所述“芳基”可为苯基、萘基、蒽基、茚基或联苯基。
所述“杂芳基”或“杂环烷基'可为氮杂环丁基、吖啶基、苯并二氧杂环戊烷基、苯并二恶烷基、苯并呋喃基、咔唑基、噌啉基、二氧戊环基、吡啶基、蝶啶基、嘌呤基、喹唑啉基、喹喔啉基、喹啉基、异喹啉基、四唑基、咪唑基、四氢异喹啉基、吡咯基、胡椒基、吡嗪基、嘧啶基、哒嗪基、吡唑啉基、恶唑基、恶唑啉基、三唑基、茚基、异恶唑基、异恶唑烷基、十氢异喹啉基、苯并咪唑基、吲唑基、苯基哌啶基、呋喃基、四氢呋喃基、四氢吡喃基、哌嗪基、高哌嗪基、哌啶基、哌立度哌啶基(Piperidoquinoline)、吗啉基、硫代吗啉基、哌啶基、2-氧代-哌嗪基、2-氧代-哌啶基、吡咯烷基、2-氧代-吡咯烷基或恶唑烷基。
所述“环烷基”可选自环丙基、环丁基、环戊基、环己基、环辛基、环庚基、全氢萘基、金刚烷基、交联的环状基团及螺环基团(spirobicyclic groups)。
在另一实施例中,所述R
Y可以为氢、4-氟苯基,或4-氯;
R
R
R
所述一个以上的取代基可以为选自4-氟、4-氯、4-羟基,以及4-三氟甲基的化合物、溶剂化物、立体异构,或其药学上可接受的盐。
在另一实施例中,由所述化学式P表示的化合物可选自:
4-氟-N-(3-(4-(6-(4-氟-3-甲氧基苯基)咪唑并[2,1-b]噻唑基-5基)嘧啶-2基氨基)丙基)苯磺酰胺(37Ⅲ);
4-氯-N-(3-(4-(6-(4-氟-3-甲氧基苯基)咪唑并[2,1-b]噻唑基-5基_)嘧啶-2基氨基)丙基)苯磺酰胺(38Ⅲ);
4-氟-N-(2-(4-(6-(4-氟-3-羟基苯基)咪唑并[2,1-b]噻唑基-5基_)嘧啶-2基氨基)乙基)苯磺酰胺(42Ⅲ);
4-氯-N-(2-(4-(6-(4-氟-3-羟基苯基)咪唑并[2,1-b]噻唑基-5基_)嘧啶-2基氨基)乙基)苯磺酰胺(43Ⅲ);
N-(2-(4-(6-(4-氟-3-羟基苯基)咪唑并[2,1-b]噻唑基-5基_)嘧啶-2基氨基)乙基)苯磺酰胺(46Ⅲ);
4-氯-N-(3-(4-(6-(4-氟-3-羟基苯基)咪唑并[2,1-b]噻唑基-5基_)嘧啶-2基氨基)丙基)苯磺酰胺(48Ⅲ);
N-(3-(4-(6-(4-氟-3-羟基苯基)咪唑并[2,1-b]噻唑基-5基_)嘧啶-2基氨基)丙基)-4-甲氧基苯磺酰胺(50Ⅲ);
N-(3-(4-(6-(3-氨基-4-氟苯基)咪唑并[2,1-b]噻唑基-5基_)嘧啶-2基氨基)丙基)-4-氟苯磺酰胺(52Ⅲ);
4-氯-N-(3-(4-(6-(4-氟-3-甲氧基苯基)咪唑并[2,1-b]恶唑-5-基)嘧啶-2-基氨基)丙基)苯磺酰胺(60Ⅳ);
4-氟-N-(2-(4-(6-(4-氟-3-羟基苯基)咪唑并[2,1-b]恶唑-5-基)嘧啶-2-基氨基)丙基)苯磺酰胺(64Ⅳ);
4-氯-N-(2-(4-(6-(4-氟-3-羟基苯基)咪唑并[2,1-b]恶唑-5-基)嘧啶-2-基氨基)乙基)苯磺酰胺(65Ⅳ);
4-氯-N-(3-(4-(6-(4-氟-3-羟基苯基)咪唑并[2,1-b]恶唑-5-基)嘧啶-2-基氨基)丙基)苯磺酰胺(70Ⅳ);
N-(3-(4-(6-(4-氟-3-羟基苯基)咪唑并[2,1-b]恶唑-5-基)嘧啶-2-基氨基)丙基)-4-甲氧基苯磺酰胺(72Ⅳ);以及
N-(3-(4-(6-(4-氟-3-羟基苯基)咪唑并[2,1-b]恶唑-5-基)嘧啶-2-基氨基)丙基)苯磺酰胺(73Ⅳ)。
根据本公开的一个方面,提供了由化学式P表示的化合物、溶剂化物、立体异构体或其药学上可接受的盐的制备方法,包括:通过将化学式1及化学式2环化来制备化学式3的化合物;
通过将化学式3的化合物与4-氯-2-(甲硫基)嘧啶进行反应来制备化学式4的化合物;
通过将化学式4的化合物氧化来制备化学式5的化合物;以及
通过将化学式5的化合物与N-N-氨乙基环磺酰胺衍生物或N-(2-氨乙基)苯磺酰胺衍生物在碱基下进行反应,来获得化学式P的化合物。
具体地,所述制备方法可以为如下反应式1所示。
[反应式1]
所述A、B及X如上所述定义。
在一个实施例中,通过将所述化学式3的化合物与4-氯-2-(甲硫基)嘧啶进行反应来制备化学式4的化合物的步骤可在赫克反应(Heck reaction)条件下进行反应,并在Pd(OAc)
根据本公开的一个方面,提供一种用于预防或治疗肿瘤的药物组合物,其包括所述化合物、溶剂化物、立体异构体或其药学上可接受的盐作为有效成分。
根据本公开的另一个方面,提供包括用于制备所述药物组合物的所述化合物、溶剂化物、立体异构体或其药学上可接受的盐的组合物的用途。
所述“药学上可接受的盐”是指所述化学式P的化合物的任何有机或无机加成盐,其中,所述任何有机或无机加成盐以对患者相对无毒且无害并具有有效作用的浓度对该盐引起的副作用不损害化学式P的化合物的有益效能。这些盐可以将无机酸和有机酸用作游离酸,可使用盐酸、溴酸、硝酸、硫酸、过氯酸和磷酸等作为无机酸。作为有机酸,可使用柠檬酸、乙酸、乳酸、马来酸、富马酸、葡萄糖酸、甲磺酸、甘醇酸、琥珀酸、酒石酸、半乳糖醛酸、扑酸、谷氨酸、天冬氨酸、草酸、(D)或(L)苹果酸、马来酸、甲磺酸、乙磺酸、4-甲苯磺酸、水杨酸、柠檬酸、苯甲酸或丙二酸等。这些盐包括碱金属盐(如钠盐和钾盐等)及碱土金属盐(如钙盐和镁盐等)等。例如,作为酸加成盐,可包括乙酸盐、天冬氨酸盐、苯甲酸盐、苯磺酸盐、碳酸氢盐/碳酸盐、硫酸氢盐/硫酸盐、硼酸盐、樟脑磺酸盐、柠檬酸盐、乙二磺酸盐、乙磺酸盐、甲酸盐、富马酸盐、葡庚糖酸盐、葡萄糖酸盐、葡糖醛酸酯、六氟磷酸盐、0-(4-羟苯甲酰)苯甲酸盐、盐酸盐/氯化物、氢溴酸盐/溴化物、氢碘酸盐/碘化物、羟乙基磺酸盐、乳酸盐、苹果酸盐、马来酸盐、丙二酸盐、甲磺酸盐、甲基硫酸盐、萘酸盐、2-萘酸盐、烟酸盐、硝酸盐、乳清酸盐、草酸盐、棕榈酸盐、扑酸盐、磷酸盐/磷酸一氢盐/磷酸二氢盐、糖酸盐、硬脂酸盐、琥珀酸盐、酒石酸盐、甲苯磺酸盐、三氟醋酸盐、铝、精氨酸、苄星、钙、胆碱、二乙胺、二乙醇胺、甘氨酸、赖氨酸、镁、甲葡胺、乙醇胺、钾、钠、氨基丁三醇及锌盐等、或其中的盐酸盐或三氟醋酸盐。
在一个实施例中,所述化合物、溶剂化物、立体异构体或药学上可接受的盐可以通过抑制蛋白激酶来抑制肿瘤细胞增殖。所述蛋白激酶与肿瘤细胞增殖有关,并可以是从V600E快速加速纤维肉瘤(RAF)、B-RAF、C-RAF、丝裂原活化蛋白激酶14(MAPK14)、FMS样的酪氨酸激酶3(FLT3)及糖原合成酶激酶3β(GSK3β)中选择的一个或更多个。
在另一实施例中,所述肿瘤可为肺癌、肝癌、食道癌、胃癌、大肠癌、小肠癌、胰腺癌、黑色素瘤、乳腺癌、口腔癌、骨癌、脑肿瘤、甲状腺癌、甲状旁腺癌、肾癌、宫颈癌、肉瘤、前列腺癌、尿道癌、膀胱癌、睾丸癌、白血病、多发性骨髓瘤、骨髓增生异常综合征或如胶质母细胞瘤的血液癌、如霍奇金病或非霍奇金淋巴瘤的淋巴瘤、白塞病、皮肤癌、牛皮癣及纤维肉瘤。
根据本公开的一个方面,提供了包括将所述化合物、溶剂化物、立体异构体或其药学上可接受的盐施用于患上肿瘤或有患上肿瘤风险的个体的预防或治疗肿瘤的方法。
所述个体可为哺乳动物,所述哺乳动物可以是人,本公开的化合物对人体的有效施用剂量可根据患者的年龄、体重、性别、剂型、健康状况和疾病程度而改变。
所述施用可以口服给药,或以各种制剂给药以用于静脉、腹腔、皮內、皮下、上皮或肌肉给药等的非口服给药,并且在进行制剂化的情况下,使用通常使用的稀释剂(诸如,填充剂、增量剂、粘合剂、润湿剂、崩解剂及表面活性剂等)或赋形剂来制备。
用于口服施用的固体制剂可包括锭剂、丸剂、散剂、颗粒剂、胶囊剂及片剂(troche),并且这种固体制剂可以通过将一种或更多种的本公开的化合物与至少一种赋形剂(诸如,淀粉、碳酸钙、蔗糖(sucrose)或乳糖(lactose)或明胶等)混合来制备。除了简单的赋形剂外,还可以使用如硬脂酸镁滑石的润滑剂。作为口服施用的液体制剂,可以使用悬浮剂、内用液剂、油剂或糖浆(syrup)剂等,并且除了常用的简单的稀释剂如水和液体石蜡外,还可以包括各种赋形剂如润湿剂、甜味剂、芳香剂及防腐剂等。
用于非口服给药的液体制剂可包括灭了菌的水溶液、非水性溶剂、悬浮剂、油剂、冷冻干燥制剂和栓剂等。作为非水性溶剂和悬浮剂,可使用丙二醇、聚乙二醇、如橄榄油的植物油及如油酸乙酯的可注射的酯等。作为栓剂的基质,可使用合成脂肪酸酯(witepsol)、聚乙二醇、乳化剂(Tween)61、可可油脂、月桂脂、甘油和明胶等。
附图说明
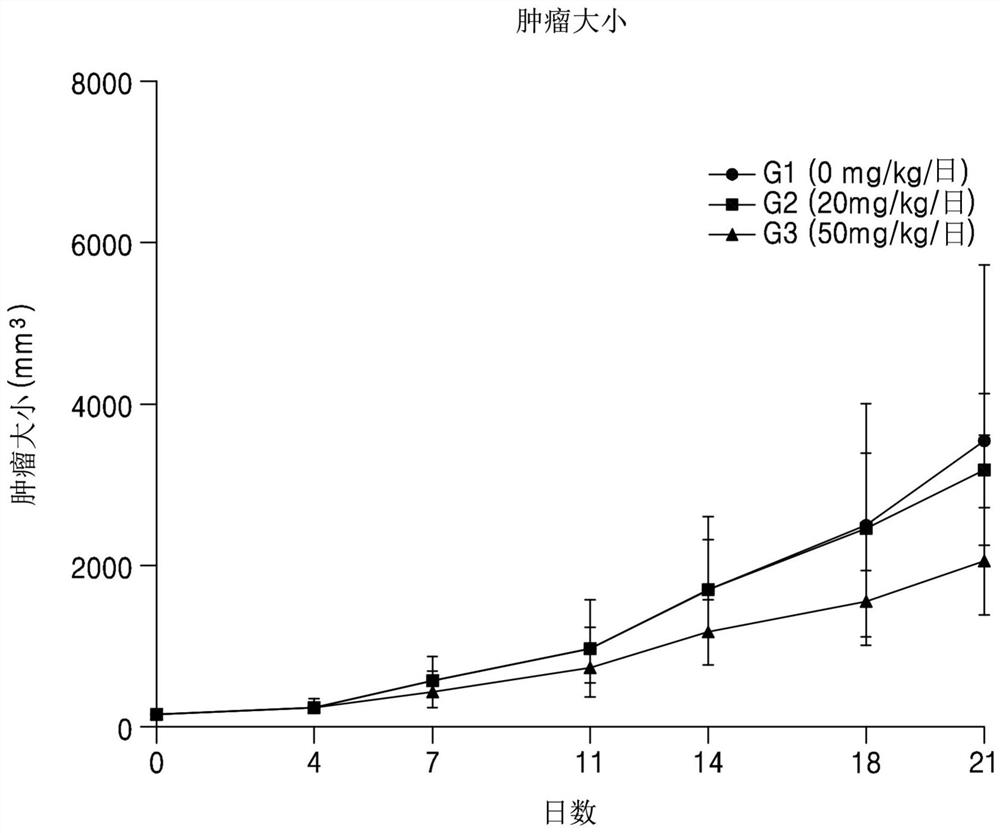
图1显示了在A375P細胞株的异种移植模型中施用化合物2Ⅱ后,根据移植后天数的肿瘤大小所表示的抗癌效果。G1表示赋形剂对照组,G2表示试验物质20mg/kg/日施用组,并且G3表示试验物质50mg/kg/日施用组。
图2是显示化合物37Ⅲ和42Ⅲ的A375P黑色素瘤细胞株增殖抑制活性的图。
具体实施方式
以下,将通过实施例更详细描述本发明。然而,这些实施例旨在以示例的方式说明本公开,并且本公开的范围不受这些实施例的限制。
如以下反应式2至反应式5中所示,根据以下工艺制备化学式I至IV的化合物。
合成咪唑并恶唑化合物的方法如下反应式2所示,为了合成所述化学式I至Ⅳ的化合物,重要的是合成作为核心中间体的化合物5。
反应式2
试剂和条件:i)乙腈(CH
通过将2-氨基恶唑(2)与α-溴-4-氟苯乙酮在乙醇下回流并反应16小时来环化,并利用氨水通过结晶化来合成化合物3。以这种方式合成的化合物3在赫克反应(Heckreaction)条件下并在Pd(OAc)
合成咪唑并噻唑基化合物的方法如下反应式3所示,除了使用2-氨基噻唑(2)之外,与反应式2相同地进行。
反应式3
试剂和条件:i)乙醇,回流;通宵ii)4-氯-2-(甲硫基)嘧啶,乙酸钯,三苯基膦,碳酸铯,DMF,80℃,16小时;iii)过氧单硫酸钾,甲醇,H
如以下反应式4和反应式5所示,根据以下工艺制备制备化学式I至IV的化合物。
反应式4
试剂和条件:ⅰ)乙醇,回流,通宵ⅱ)4-氯-2-(甲硫基)嘧啶,乙酸钯,三苯基膦,碳酸铯,DMF,80℃,16小时;ⅲ)过硫酸氢钾复合盐,甲醇,H
合成咪唑并噻唑基化合物的方法如下反应式5所示,除了使用2-氨基噻唑(2)之外,与反应式4相同地进行。
反应式5
试剂和条件:ⅰ)乙醇,回流;通宵ⅱ)4-氯-2-(甲硫基)嘧啶,乙酸钯,三苯基膦,碳酸铯,DMF,80℃,16小时;ⅲ)过硫酸氢钾复合盐,甲醇,H
每个步骤的化合物显示在以下实施例中。
实施例1.6-苯基咪唑并[2,1-b]恶唑(反应式2中的3a)
将2-氨基恶唑(反应式2中的2,4.7g,47.4mmol)和α-溴-4-苯乙酮(10.8g,49.9mmol)加入到乙腈(120mL),并在室温下搅拌48小时。在使用薄层色谱(TLC)确认反应结束后,进行过滤。将生成的固体放入甲苯(100mL)中。在0℃下缓慢加入20mL的氯化钛(1M的二氯甲烷溶液)到悬浮液中。将反应溶液回流并搅拌20小时。将以上溶液浓缩至其原始体积的一半。在加入饱和碳酸钠溶液后,使用乙酸乙酯进行提取。用无水硫酸钠Na
白色固体(5.1g,56%),熔点143℃至144℃;IR(KBr)[cm
实施例2.5-(2-(甲硫基)嘧啶-4-基)-6-苯基咪唑并[2,1-b]恶唑(反应式2中的4a)
将6-(4-苯基)咪唑并[2,1-b]恶唑(所述化合物3a,8.26g,37.8mmol)、4-氯-2-(甲硫基)嘧啶(9.09g,56.6mmol)、碳酸铯(18.44g,56.6mmol)、乙酸钯(1.71g,7.6mmol)及三苯基膦(3.96g,15.1mmol)加入到DMF(80mL)中,并在80℃下搅拌36小时。在用TLC确认反应结束后,冷却至室温,并加入乙酸乙酯(EtOAc)(200mL)和蒸馏水(250mL)并对层进行分离。在用EtOAc(100mL)提取3次水层后,丢弃水层。用盐水(saline)(100mL)洗涤有机层,并用无水Na
白色固体(19%),熔点143℃至144℃;IR(KBr)[cm
实施例3.5-(2-(甲磺酰基)嘧啶-4-基)-6-苯基咪唑并[2,1-b]恶唑e(反应式2中的5a)
将6-(4-苯基)-5-(2-(甲硫基)嘧啶-4-基)咪唑并[2,1-b]恶唑(所述化合物4a,3.0g,8.8mmol)加入到甲醇(MeOH)(300mL)中并在室温下搅拌的同时,加入蒸馏水(60L)中的过氧单硫酸钾(14.76g,97.0mmol)并搅拌16小时。在用TLC确认反应结束后,通过减压蒸馏除去有机溶剂。将二氯甲烷(DCM)(60mL)加入到水层中来对层进行分离。在用DCM(30mL)提取三次水层后,丢弃水层。在用盐水(50mL)和蒸馏水(50mL)洗涤有机层后,并用无水Na
白色固体(85%);
实施例4.6-(4-氟苯基)-5-(2-(甲磺酰基)嘧啶-4-基)咪唑并[2,1-b]恶唑(反应式2中的5c)
通过与所述化合物5a相同的实验方法来进行合成。
黄色固体(85%);熔点.115℃至116℃.
实施例5.5-(2-(甲磺酰基)嘧啶-4-基)-6-苯基咪唑并[2,1-b]噻唑(反应式3中的5d)
通过与所述化合物5a相同的实验方法来进行合成。
白色固体(95%);熔点.176℃至177℃.IR(KBr)[cm
实测值6.2-甲基-1,2,5-噻二唑烷1,1-二氧化物(6a)
将N-甲基乙二胺(1.5g,20.8mmol)和硫二亚胺(2.0g,20.8mmol)加入到吡啶溶剂(20mL)中,并回流搅拌加热3小时。在反应结束后将反应物加入到甲苯(5mL)中并进行浓缩后,使用乙酸乙酯进行提取,并通过使用硫酸镁(MgSO
白色固体(52.3%).熔点80℃至82℃;
实施例7.苄基(2-(5-甲基-1,1-二氧化物-1,2,5-噻二唑烷-2-基)乙基)氨基甲酸酯(7a)
在氮气气氛下,将所述化合物6a(0.856g,6.26mmol)溶于5mL的无水DMSO后,在0℃下缓慢滴加60%氢化钠(NaH)(0.282g,11.8mmol)。在升温至室温之后,搅拌1小时。在0℃下缓慢滴加2-((苄氧基)羰基)氨基)乙基甲磺酸盐(1.5g,5.83mmol)后,在室温下搅拌5小时。在反应结束后,用乙酸乙酯进行提取,并用MgSO
有粘性的油(42.9%).
实施例8.在2-(2-氨乙基)-5-甲基-1,2,5-噻二唑烷1,1-二氧化物(8a)的合成(Synthesis)
将所述化合物7a(0.82g,2.5mmol)溶解在10mL的甲醇后,加入10%钯碳催化剂(Pd/C)(0.40g)并通过加氢反应反应2小时,进行过滤,并通过减压蒸馏来除去溶剂。不进行纯化,并用于后续反应(收率91%)。
实施例9.N-(2-氨乙基)-4-氟苯磺酰胺(11b)
白色固体(72%);熔点.108℃至109℃;IR(KBr)cm
实施例10.2-(2-((4-(6-(4-氟苯基)咪唑并[2,1-b]恶唑-5-基)嘧啶-2-基)氨基)乙基)-5-甲基-1,2,5-噻二唑烷1,1-二氧化物(1Ⅰ)
淡黄色固体;在5mL的DMSO加入化合物6-(4-氟苯基)-5-(2-(甲磺酰基)嘧啶-4-基)咪唑并[2,1-b]噻唑(化合物5c,0.19g,0.50mmol)和2-(2-氨乙基)-5-甲基-1,2,5-噻二唑烷1,1-二氧化物(化合物8a,0.27g,1.24mmol),以及DIPEA(0.29mL,1.70mmol),并在80℃下搅拌8小时。将所述混合物冷却至室温,并加入蒸馏水(10mL)和EtOAc(10mL)以对层进行分离。用EtOAc(10mL)提取两次水层并弃去水层。用盐水(10mL)洗涤有机层。用无水硫酸钠Na
(72%);
实施例11.2-(2-((4-(6-(3-甲氧基苯基)咪唑并[2,1-b]恶唑-5-基)嘧啶-2-基)氨基)乙基)-5-甲基-1,2,5-噻二唑烷1,1-二氧化物(4Ⅰ)
通过与所述化合物1I相同的实验方法来进行合成。
白色固体(50.5%);熔点144℃至145℃;
实施例12.2-(2-((4-(6-(3-羟基苯基)咪唑并[2,1-b]恶唑-5-基)嘧啶-2-基)氨基)乙基)-5-甲基-1,2,5-噻二唑烷1,1-二氧化物(1Ⅱ)
在氮气环境下,将化合物4I(17.5mg,0.1mmol)溶解在3mL的MC中,并在-78℃下滴加BBr
白色固体(78.1%);熔点151℃至152℃;
实施例13.2-乙基-5-(2-((4-(6-(3-羟基苯基)咪唑并[2,1-b]恶唑-5-基)嘧啶-2-基)氨基)乙基)-1,2,5-噻二唑烷1,1-二氧化物(2Ⅱ)
通过与所述化合物1Ⅱ相同的实验方法进行合成。
白色固体(66.5%);熔点149℃至150℃;
实施例14.2-(2-((4-(6-(3-羟基苯基)咪唑并[2,1-b]恶唑-5-基)嘧啶-2-基)氨基)乙基)-4,4-甲基-1,2,5-噻二唑烷1,1-二氧化物(5Ⅱ)
通过与所述化合物1Ⅱ相同的实验方法进行合成。
白色固体(25.0%);熔点196℃至197℃;
实施例15.2-(2-((4-(6-(3-羟基苯基)咪唑并[2,1-b]恶唑-5-基)嘧啶-2-基)氨基)乙基)-6-甲基-1,2,6-噻二唑烷1,1-二氧化物(6Ⅱ)
通过与所述化合物1Ⅱ相同的实验方法合成。
白色固体(71.7%);熔点184℃至185℃;
实施例16.2-(2-((4-(6-(3-羟基苯基)咪唑并[2,1-b]恶唑-5-基)嘧啶-2-基)氨基)乙基)-1,2,6-噻二唑烷1,1-二氧化物(8Ⅱ)
通过与所述化合物1Ⅱ相同的实验方法进行合成。
白色固体(85.3%);熔点183℃至184℃;
实施例17.4-氟-N-(2-((4-(6-苯基咪唑并[2,1-b]噻唑基-5-基)嘧啶-2-基)氨基)乙基)苯磺酰胺(1Ⅲ)
在氮气环境下,将化合物5d(327mg,0.92mmol)、N-(3-氨乙基)-4-氟苯磺酰胺(11b,540mg,2.48mmol)及二异丙基乙胺(DIPEA)(0.57mL 3.3mmol)加入到DMSO溶剂(10mL)后,在80℃下搅拌8小时。当反应结束时,用乙酸乙酯进行提取,并用MgSO
橙色固体(52.1%);熔点.121℃至122℃;
实施例18.4-甲氧基-N-(2-((4-(6-苯基咪唑并[2,1-b]噻唑-5-基)嘧啶-2-基)氨基)乙基)苯磺酰胺(2Ⅲ)
通过与所述化合物1Ⅲ相同的实验方法进行合成。
白色固体(65%);熔点.199℃至200℃;
实施例19.N-(2-((4-(6-(3-甲氧苯基)咪唑并[2,1-b]噻唑-5-基)嘧啶-2-基)氨基)乙基)-4-(三氟甲基)苯磺酰胺(8Ⅲ)
通过与所述化合物1Ⅲ相同的实验方法进行合成。
无色有粘性的油(25%);
实施例20.4-氟-N-(2-((4-(6-(3-羟基苯基)咪唑并[2,1-b]噻唑-5-基)嘧啶-2-基)氨基)乙基)苯磺酰胺(25Ⅲ)
通过与所述化合物1Ⅱ相同的实验方法进行合成。
深橙色固体(44%);熔点70℃至71℃;IR(KBr)[cm
实施例21.4-氟-N-(3-((4-(6-(3-羟基苯基)咪唑并[2,1-b]噻唑-5-基)嘧啶-2-基)氨基)丙基)苯磺酰胺(31Ⅲ)
通过与所述化合物1Ⅱ相同的实验方法进行合成。
淡黄色固体(20%);熔点92℃至93℃;IR(KBr)[cm
实施例22.6-(4-氟-3-甲氧苯基)咪唑并[2,1-b]噻唑基(反应式5中的3i)
将2-溴-1-(4-氟-3-甲氧苯基)乙-1-醇(44.6mmol)和2-氨基噻唑(5,4.46g,44.6mmol,1Eq)的混合物溶于无水乙醇18小时,并搅拌回流。将反应混合物在减压下浓缩。加入冰冷的水(Ice-cold water)(50ml),然后加入氨溶液(30%,100ml)。将反应混合物在室温搅拌2小时。滤出形成的沉淀物,并用水(2x20mL)洗涤且干燥来获得固体产物。产物通过柱色谱法纯化。
实施例23.6-(4-氟-3-甲氧苯基)-5-(2-(甲硫基)嘧啶-4-基)咪唑并[2,1-b]噻唑基(反应式5中的4j)
在三颈烧瓶(three neck flask)中,将4-氯-2-(甲硫基)嘧啶(7,70.7mg,0.44mmol,1Eq),柠檬酸钾(60.8mg,0.44mmol,1Eq),乙酸钯(19.8mg,0.09mmol,0.2Eq),和三苯基膦(34.6mg,0.13mmol,0.3Eq)与化合物6-(4-氟-3-甲氧苯基)咪唑并[2,1-b]噻唑基(0.44mmol,1Eq)混合。用氮气代替空气。加入无水DMF(10ml),混合物用氮气清洗(purged)几次。将反应混合物在80℃下搅拌18小时。将反应混合物冷却,并在EA(20ml)和水(10ml)之间提取。分离有机层,用无水硫酸钠干燥并蒸发。残余物无需进一步纯化即可用于下一步骤。
实施例24.6-(4-氟-3-甲氧苯基)-5-(2-(甲磺酰基)嘧啶-4-基)咪唑并[2,1-b]噻唑基(反应式5中的5j)
在室温下,在甲醇(10ml)中的6-(4-氟-3-甲氧苯基)5-(2-(甲磺酰基)嘧啶-4-基)咪唑并[2,1-b]噻唑基(0.44mmol)的溶液中逐滴添加水(10ml)中的过硫酸氢钾复合盐(0.9g,1.32mmol)溶液。将混合物在室温搅拌48小时。将反应混合物在减压下浓缩。反应物用二氯甲烷(20ml)提取,分离有机层。用二氯甲烷(3×10ml)提取水层。合并的有机层用无水硫酸钠干燥,并减压蒸发。残留物通过柱色谱法纯化。
实施例25.4-氟-N-(3-((4-(6-(4-氟-3-甲氧苯基)咪唑并[2,1-b]噻唑-5-基)嘧啶-2-基)氨基)丙基)苯磺酰胺(37Ⅲ)
在DMSO(3ml)中的6-(4-氟-3-甲氧苯基)-5-(2-(甲磺酰基)嘧啶-4-基)咪唑并[2,1-b]噻唑基(0.26mmol)的溶液中逐滴添加N-(3-氨丙基)-4-氟苯磺酰胺(0.39mmol)和DIPEA(300mg,2.34mmol)。将反应混合物在100℃下搅拌18小时。将反应混合物冷却,并在EA(20ml)和水(10ml)之间提取。用无水硫酸钠干燥有机層,并在减压下蒸发。残留物通过柱色谱法纯化。
收率:70%.1H NMR(400MHz,DMSO-d6)δ8.08(d,J=8.0Hz,1H),7.88(q,J=4.0Hz,3H),7.69(s,1H),7.43-7.37(m,4H),7.22(t,J=4.0Hz,1H),6.54(s,1H),3.86(s,3H),2.91(d,J=8.0Hz,2H),2.52(s,2H),1.81(d,J=4.0Hz,2H).LC/MS 558(M++1).
实施例26.4-氟-N-(3-((4-(6-(4-氟-3-羟基苯基)咪唑并[2,1-b]噻唑-5-基)嘧啶-2-基)氨基)丙基)苯磺酰胺(42Ⅲ)
在-78℃在氮气下,在二氯甲烷(5mL)中的4-氟-N-(3-((4-(6-(4-氟-3-羟基苯基)咪唑并[2,1-b]噻唑基-5-基)嘧啶-2-基)氨基)丙基)苯磺酰胺(0.1mmol)的混合物中滴加BBr
收率:40%.1H NMR(400MHz,MeOD)δ8.78(s,1H),8.03(d,J=5.2Hz,1H),8.89(q,J=5.2Hz,4H),7.34-7.24(m,5H),7.19-7.15(m,2H),7.05-7.01(m,1H),6.45(d,J=5.6Hz,1H),3.02(t,J=6.8Hz,2H),2.89(t,J=6.8Hz,2H),1.83(t,J=6.8Hz,2H.LC/MS 543(M++1).
实施例27.6-(3-硝基苯基)咪唑并[2,1-b]噻唑基(反应式5中的3l)
将MeOH(50mL)中的化合物1i(1g,4.1mmol,1Eq)和化合物2(0.5g,4.9mmol,1.2Eq)的溶液在回流下搅拌18小时。真空蒸发有机溶剂。将固体沉淀物在NH
收率:90%.m.p.:167-9℃.1H NMR(400MHz,DMSO-d6)δ8.63(t,J=2.2Hz,1H,Ar-H),8.27-8.24(m,1H,Ar-H),8.09-8.06(m,1H,Ar-H),7.98(d,J=4.4Hz,1H,Ar-H),7.67(t,J=8.2Hz,1H,Ar-H),7.32(d,J=4.4Hz,1H,Ar-H).13C NMR(100MHz,DMSO-DMSO-d6)δ150.25(Ar-C),148.78(Ar-C),144.40(Ar-C),136.46(Ar-C),131.26(Ar-C),130.61(Ar-C),121.87(Ar-C),120.52(Ar-C),119.32(Ar-C),114.39(Ar-C),111.44(Ar-C).
实施例28.5-(2-(甲硫基)嘧啶-4-基)-6-(3-硝基苯基)咪唑并[2,1-b]噻唑基(反应式5中的4l)
将无水DMF(5mL)中的溶液化合物6-(3-硝基苯基)咪唑并[2,1-b]噻唑基(0.7g,4.1mmol,1Eq)滴加到无水DMF(10mL)中的氯甲硫基嘧啶(1g,4.1mmol,1Eq),Ph3P(0.3g,1.3mmol,0.3Eq),K2CO3(0.6g,4.1mmol,1Eq),以及Pd(OAc)2(0.2g,0.8mmol,0.2Eq)的混合物中。将反应混合物在80℃下搅拌8小时。将反应混合物冷却并与碎冰(30g)一起搅拌。滤出沉淀物。将固体残余物在60℃下在MeOH(50ml)中搅拌1小时。滤出固体产物,用MeOH(3×20ml)洗涤且干燥,得到标题化合物。
收率:30%.m.p.:187-9℃.1H NMR(400MHz,CDCl3)δ8.58(d,J=4.4Hz,2H,Ar-H),8.34-8.31(m,2H,Ar-H),8.02(d,J=8.0Hz,1H,Ar-H),7.66(t,J=8.0Hz,1H,Ar-H),7.06(d,J=4.4Hz,1H,Ar-H),6.86(d,J=5.6Hz,1H,Ar-H),2.66(s,3H,SCH3).13C NMR(100MHz,CDCl3)δ173.15(Ar-C),156.83(Ar-C),155.66(Ar-C),153.01(Ar-C),148.57(Ar-C),147.65(Ar-C),136.32(Ar-C),135.04(Ar-C),129.83(Ar-C),124.14(Ar-C),123.53(Ar-C),121.96(Ar-C),120.82(Ar-C),113.65(Ar-C),112.00(Ar-C),14.23(SCH3).LC/MS 371(M+1)+.
实施例29.5-(2-(甲硫基)嘧啶-4-基)-6-(3-硝基苯基)咪唑并[2,1-b]噻唑基(反应式5中的5l)
将水(20mL)中的过氧单硫酸钾盐(5g,8.1mmol,3Eq)的溶液滴加到MeOH(50ml)中的5-(2-甲硫基)嘧啶-4-基)-6(3-硝基苯基)咪唑并[2,1-b]噻唑基(1g,2.7mmol,1Eq)的溶液中。将反应混合物在室温搅拌9小时。真空蒸发有机溶剂。将残余物在DCM(70ml)和水(30ml)之间提取。有机层用盐水溶液(3×30ml)洗涤。有机层经无水Na
收率:80%.m.p.:215-6℃.1H NMR(400MHz,CDCl3)δ8.91(d,J=4.4Hz,1H,Ar-H),8.61(t,J=5.6Hz,2H,Ar-H),8.39-8.37(m,1H,Ar-H),8.04(d,J=8.0Hz,1H,Ar-H),7.75(t,J=7.6Hz,1H,Ar-H),7.35(d,J=5.6Hz,1H,Ar-H),7.17(d,J=4.8Hz,1H,Ar-H),3.42(s,3H,SO
实施例30.N-(3-((4-(6-(3-氨基-4-氟苯基)咪唑并[2,1-b]噻唑-5基_)嘧啶-2-基)氨基)丙基)-4-氟苯磺酰胺(52Ⅲ)
在DMSO(3mL)中的6-(4-氟-3-硝基)-5-(2-(甲磺酰基)嘧啶-4-基)咪唑并[2,1-b]噻唑基(0.26mmol)的溶液中加入N-(3-氨丙基)-4-氟苯磺酰胺(0.39mmol)和DIPEA(300mg,2.34mmol)。将反应混合物在100℃下搅拌18小时。将反应混合物冷却,并在EA(20ml)和水(10ml)之间提取。有机层经无水硫酸钠干燥,并在减压下蒸发。将残余物溶于10ml甲醇中,并加入10%pd/C,并将混合物在氢气氛下通宵搅拌。反应完成后,滤出pd/C,并蒸发甲醇。使用柱色谱法纯化残余物。
1H NMR(400MHz,MeOH)δ8.74(s,1H),7.98(d,J=4.0Hz,2H),7.91-7.87(m,2H),7.31-7.23(m,3H),7.09-7.03(m,2H),6.83-6.79(m,1H),6.45(d,J=4.0Hz,1H),4.47(s,2H),3.01(t,J=4.0Hz,2H),1.81(t,J=8.0Hz,2H).13C NMR(100MHz,MeOH)δ166.23,163.01,162.01,156.25,153.24,151.64,150.01,149.11,136.23,130.11,129.78,120.12,118.15,117.12,115.52114.81,105.91,40.35,38.19,29.1
除了使用不同的取代基之外,参考上述合成方法以相同方式合成其他化合物。下面的表1显示了由此合成化合物的例子。
【表1】
<实验例1>测量黑色素瘤细胞株增殖的抑制活性
为了测量根据本公开的咪唑并恶唑类或咪唑并噻唑类化合物在细胞阶段中的癌细胞增殖抑制活性,进行了以下实验。
根据美国国家癌症研究所(National Cancer Institute,NCI)(www.dtp.nci.nih.gov)的标准协议(http://dtp.nci.nih.gov/dtpstandard/dwindex/index.jsp),执行关于癌细胞株组(panel)的筛选。简而言之,使人细胞株在包括5%胎牛血清(Fetal Bovine Serum,FBS)及L-谷氨酰胺的RPMI 1640培养基中生长。为了典型的筛选实验,将细胞按照100μl接种到96孔微量滴定板(96-well microtiter plate)中,以使根据单独的细胞株的倍增时间(doubling time),以范围为5000个细胞/孔至40,000个细胞/孔的浓度进行平板接种。在细胞接种后,在添加测试化合物之前,在37℃、5%CO
加入化合物后,将所述滴定板在37℃、5%二氧化碳、95%空气以及100%相对湿度下进一步培养48小时。在贴壁细胞的情况下,通过加入冷TCA来结束所述分析。通过温和地加入冷50%的50μl(w/v)的TCA(最终浓度10%的TCA),以将细胞固定在该状态,并在4℃下培养60分钟。弃去上清液,并用自来水将所述板洗涤5次并进行空气干燥。向每个孔中加入在1%乙酸中0.4%(w/v)的磺酰罗丹明B(SRB)溶液(100μl),并将滴定板在室温下培养10分钟。染色后,用1%乙酸洗涤5次以除去未结合的染料,对所述滴定板进行空气干燥。将结合的染料依次用10mM的三羟甲基氨基甲烷碱溶解,并用自动化平板读数器在515nm波长下读取吸光度。对于悬浮细胞,该方法是相同的,除了通过温和地加入50μl的80%TCA(最终浓度16%TCA)以固定沉淀在孔的底部的细胞来终止所述分析。使用七种吸光度的测量[在零点时间(Tz)、对照组生长率(C)及五种浓度等级下存在化合物的情况下的测试生长率(Ti)],在每个化合物浓度等级下计算生长百分率。生长抑制百分率的计算如下。
对于Ti>/=Tz的浓度[(Ti-Tz)/(C-Tz)]x 100
对于Ti 表2对于在体外黑色素瘤细胞株的主要化合物的IC 【表2】
在所述表2中所示的大多数黑色素瘤细胞株中,显示了试验化合物具有增殖抑制活性(均小于10μM),由此可以看出,根据本发明的咪唑并恶唑类化合物或咪唑并噻唑类化合物对黑色素瘤具有优异的抑制效果。
与本发明人先前公开(M.S.阿卜杜勒-马克苏德(Abdel-Maksoud)等人./欧洲药物化学期刊(European Journal of Medicinal Chemistry)95(2015)453-463)的咪唑并噻唑类化合物相比较,为了确认本发明的化合物(2Ⅱ、9Ⅱ、10Ⅱ及12Ⅱ至15Ⅱ)是否具有更显著的作用,使用作为黑色素瘤细胞株的A375P细胞株进行如下的增殖抑制活性的比较实验。 将从ATCC购买的A375P细胞株在存在5%二氧化碳的情况下于37℃在DMEM培养液(包括10%FBS、1%青霉素/链霉素(penicillin/streptomycin))中培养。将培养的A375P细胞株用0.05%胰蛋白酶(trypsin)-0.02%乙二胺四乙酸(EDTA)取出,以在每孔(well)5×10 表3主要咪唑并噻唑类化合物的A375P细胞株增殖抑制活性的IC 【表3】
如所述表3和图2所示,相比现有的咪唑并噻唑类化合物P1至P5,可以看出本公开的化合物2Ⅱ、9Ⅱ、10Ⅱ及12Ⅱ至15Ⅱ具有整体更好的增殖抑制效果,当比较相似的结构时(化合物P1/9Ⅱ、P2/10Ⅱ、P3/2Ⅱ及P4/12Ⅱ),可以更清楚地看出,本公开的化合物具有更显著的效果。特别是,如图2所示,结果显示当所述化学式P的A具有两个取代基R <实验例2>测量蛋白激酶的酶活性 通过以下方法得知根据本公开的咪唑并恶唑类及咪唑并噻唑类化合物关于黑色素瘤细胞株的增殖抑制活性是否抑制激酶的酶活性。 用反应生物激酶热点服务(Reaction Biology Kinase Hotspot Service)(http://www.reactionbiology.com)测量了IC 表4对于B-RAF、V600E-RAF及C-RAF的激酶活性抑制效果(nM) 【表4】
如所述表4所示,在根据本公开的化合物中的化合物1Ⅱ、2Ⅱ、25Ⅲ及31Ⅲ的情况下,对于B-RAF、V600E-RAF及C-RAF的酶活性抑制效果显示出优异,并且可得知比GW5074和维罗非尼(Vemurafenib)优异。特别是,当所述化学式P的A具有两个取代基R 总之,由于根据本公开的化合物表现出对蛋白激酶,特别是引起如肿瘤细胞的异常细胞生长疾病的各种蛋白激酶的优异抑制活性,如对于V600E RAF、B-RAF、C-RAF、MAPK14、FLT3及GSK3β显示出优异的抑制效果,因此显示出可用于预防和治疗肿瘤细胞生长疾病。 <实验例3>在黑色素瘤小鼠模型中的肿瘤抑制效果 为了得知在动物模型中能否再现在细胞水平上看到的黑色素瘤细胞增殖抑制效果和对于所述B-RAF、V600E-RAF及C-RAF的酶活性抑制效果,进行了如下试验。 在本试验中,通过将源于人的恶性黑色素瘤(malignant melanoma)细胞株的A375P细胞株皮下施用于裸鼠来制作异种移植模型之后,施用测试物质(化合物2Ⅱ)以评估抗癌效果。(G1:赋形剂对照组,G2:试验物质20mg/kg/日施用组,G3:试验物质50mg/kg/日施用组) 结果显示在图1和以下表5中。 表5在黑色素瘤异种移植模型的肿瘤大小的增加表(2Ⅱ化合物) 【表5】
一般症状的观察结果未观察到死亡动物或一般症状的观察结果方面的异常。作为体重测量的结果,观察到在试验物质施用后第14天和第21天,试验物质50mg/kg/日施用组(G3)的体重比对照组(G1)显著地低,并且试验物质50mg/kg/日施用组的体重增加显著低于赋形剂对照组。 虽然肿瘤大小测量结果未显示所有试验组之间的任何显著差异,但在试验物质施用组中观察到存在与剂量相关地抑制肿瘤大小增加的趋势。 总之,当将试验物质以两次/周持续3周静脉施用于使用裸鼠的恶性黑色素瘤异种移植模型时,虽然试验物质施用组的体重比对照组有明显低或低的趋势,并观察到体重增加减小,但观察到存在以剂量相关地抑制肿瘤大小增加的趋势。因此,由于已经显示公开的化合物不仅能够在细胞水平上抑制肿瘤生长,而且能够在动物试验中抑制肿瘤生长,因此可确定所述化合物具有治疗和预防肿瘤的效果。
- 具有抗肿瘤效果的咪唑并恶唑衍生物及包括其的药物组合物
- 具有抗肿瘤效果的咪唑并恶唑衍生物及包括其的药物组合物