用作可再充电锂离子蓄电池的正电极材料的前体的钴氧化物
文献发布时间:2023-06-19 12:02:28
技术领域和背景技术
本文描述了用于基于可再充电层状锂钴氧化物的蓄电池中的正电极活性材料的钴氧化物前体粉末。所述钴氧化物前体粉末包含具有相对较高机械强度以及相对较大平均粒度的颗粒,这可有利于为具有由该钴氧化物前体粉末制备的正电极活性材料的蓄电池产生高能量密度和循环能力。该钴氧化物前体粉末可通过在第一干燥非氧化气氛和第二干燥氧化气氛下的连续加热工艺步骤来制备。
基于层状锂钴氧化物的可再充电蓄电池作为一种(可再充电)锂离子蓄电池(LIB)由于其高体积和重量能量密度以及长循环寿命,目前被用于膝上型电脑、移动电话、相机和多种其它便携式电子设备。此外,层状LiCoO
此外,为了满足对便携式应用日益增长的需求,需要具有较高能量密度的蓄电池。
为了制备LCO,通常使用基于钴的前体,诸如例如碳酸钴(CoCO
在一般制备过程中,将钴前体与诸如例如碳酸锂(Li
便携式应用的一个重要蓄电池特性在于蓄电池的能量密度。能量密度确定储存电能的蓄电池尺寸。存在两种类型的能量密度:重量能量密度和体积能量密度。蓄电池中正电极材料的类型决定蓄电池中的能量密度。由于消费者对较小电子设备的需求,体积能量密度在便携式应用中尤其重要。因此,用于便携式电子设备中的蓄电池包含具有较高体积粉末密度的正电极材料是有利的。可以通过向正电极材料施加压力来增加体积能量密度。然而,向正电极材料施加过大的压力可对放置材料的蓄电池的循环寿命产生有害影响。因此,期望一种用于改善体积能量密度的可行的替代选择。
本文所述的钴前体可解决上述问题,而从现有技术例如从US2017/062807(下文称为“US'807”)、US2016/322633(“US'633”)、US2012/134914(“US'914”)、US2007/099087(“US'087”)、WO2018/162165(“WO'165”)和WO2018/052210(WO'210)中已知的钴前体则反之。例如,由所述钴前体粉末制备的正电极材料可具有高体积密度以及更长的循环寿命,同时易于制造,这意味着由所述钴前体粉末制备的正电极材料可在标准烧结条件下获得。
发明内容
本发明涉及以下实施方案:
更优选地,在该实施方案1中,该钴氧化物前体粉末具有圆度大于或等于0.80并且小于或等于1.00的颗粒。
根据前述实施方案中任一项所述的实施方案4:在第一方面的另一个特征中,该粉末中颗粒的摩尔比Li/(Co+A)变化小于3%。
在该方面的另一个特征中,在干燥惰性气氛中(例如,在干燥氮气或氮基气氛中)的加热进行约2-4小时、约2.5-3.5小时或约3小时。在又一个特征中,在干燥氧化气氛中的加热进行约2-4小时、约2.5-3.5小时或约3小时。该干燥氧化气氛可包括空气。
在该方面的特征中,根据涉及第一方面的前述实施方案中任一项所述的前体粉末与所述含Li前体粉末以Li/(Co+A)>1.00的摩尔比混合。
-在干燥惰性气氛中,优选地在干燥氮气或氮基气氛中,将包含钴盐颗粒的组合物于约300℃至约450℃的温度下加热约2-5小时,以形成中间体钴前体颗粒,以及
-在干燥氧化气氛中,将该中间体前体颗粒于约300℃至约600℃的温度下加热约2-5小时,以形成该钴氧化物前体粉末。
在本发明的框架中,于300℃至600℃的温度下在干燥(空气、氧化、氮气或N
附图说明
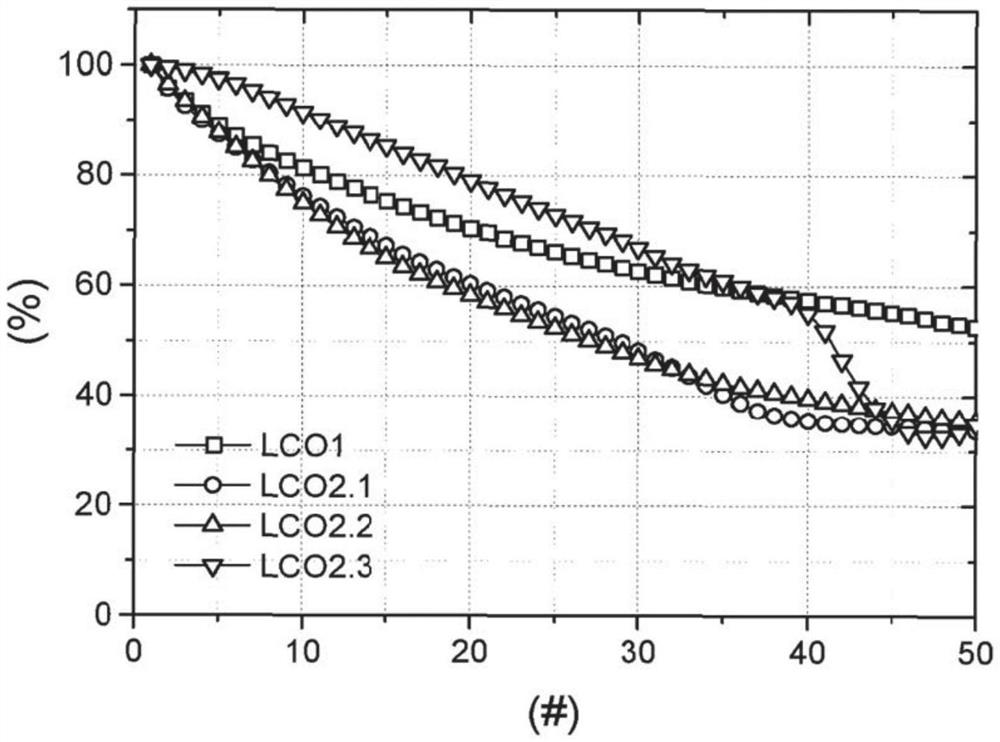
图1为示出用作循环次数函数的LCO1和LCO2.1至LCO2.3的剩余容量的图表,其中x轴为循环次数,并且y轴为剩余容量。
图2A是示出EX1和LCO1的PSD曲线的图表,其中x轴为粒度,并且y轴为体积。
图2B为示出CEX1.1和LCO2.1的PSD曲线的图表,其中x轴为粒度,并且y轴为体积。
图3为示出在锂化后在托盘中选择用于ICP分析的示例性LCO产品颗粒的各种位置的示意图。
图4为示出在图3所示的托盘中选择的LCO产品的Li/M'比率的图表。
具体实施方式
在附图和以下具体实施方式中,对实施方案进行了详细描述以实践本发明。尽管参照这些特定实施方案描述了本发明,但应当理解,本发明不限于这些实施方案。相反,本发明包括许多替代、修改和等同形式,通过考虑以下具体实施方式和附图,这些形式将变得显而易见。
本文所述的Fd-3m钴前体粉末由式Co
使用根据本发明的钴前体粉末作为LCO化合物的前体具有多种益处和优点。使用根据本发明的钴前体制备的LCO化合物的循环寿命和能量密度比较优越。另外,由于根据本发明的前体的钴含量高,LCO制备的成本降低,因为就基于氢氧化物或碳酸盐的前体而言对于给定的前体体积/质量,前体氧化物允许更高的Co负载。
该钴前体粉末的物理特性使得能够改善使用所述钴前体粉末制备的LCO化合物的循环寿命。实现该成果是因为由根据本发明的前体粉末制备的LCO化合物保持D50≥15μm,并且所述前体的压缩强度为至少100MPa且至多170MPa。例如,该钴前体粉末具有有利的高压缩强度(即,至少100MPa)。在LCO化合物的大批量制备过程中,钴前体以及LCO化合物连续地承受机械应力。例如,制备过程中的第一步是在具有高混合能量的工业混合器中将钴前体与锂源(例如,Li
然而,具有高于170MPa的压缩强度的钴前体粉末是不可取的。该钴前体颗粒通常是致密且无内部孔。该钴前体和锂源在烧结过程期间(在所述前体的锂化步骤处)的反应相对较慢,因为锂离子应迁移通过该钴前体结晶良好的块体。因此,当该钴前体具有高于170MPa的压缩强度时,推荐更高的烧结温度或更长的烧结时间,造成由具有高于170MPa的压缩强度的钴前体的锂化产生的正极材料的制造过程昂贵且不可行。另外,由所述前体粉末制备的LCO化合物,其D50≥15μm并且压缩强度为至少100MPa且至多170MPa,相对于不具有此类尺寸和强度特征的LCO化合物具有优越的体积密度,从而改善能量密度。
钴盐前体,诸如碳酸钴前体(例如,CoCO
此外,正电极材料表面上的掺杂或涂层可提供高容量和改善的稳定性。例如,掺杂的LiCo
此外,诸如CoO或Co
如以下示例所示,当使用相同工艺制备LCO化合物时,使用粒度小于15μm的钴氧化物前体制备的LCO化合物的性能比使用粒度为至少15μm的钴氧化物前体制备的LCO化合物的性能差。
在示例性实施方案中,钴氧化物前体可如下制备:
以引用方式并入本文的美国专利No.9,972,843中所述的沉淀方法可用于制备平均粒度为至少15μm的CoCO
具有所需尺寸的沉淀原料(例如,CoCO
原料的平均粒度和焙烧过程中的条件影响钴氧化物前体的物理特性,并因此影响由此得到的LCO化合物的电化学特性。就粒度而言,在沉淀期间制备的原料颗粒的平均粒度确定所得钴氧化物的平均粒度,因为颗粒的形态在焙烧之后保持相对不变。然而,在焙烧之后可预期粒度有部分收缩。
关于电化学性能,钴氧化物前体的机械强度取决于焙烧过程中的操作条件。具体地讲,如将在下面的示例中示出,当原料先在非氧化气氛然后在氧化气氛中焙烧(或在干燥气氛中加热或烧结)时,所得钴氧化物的机械强度高。在示例性实施方案中,当在焙烧过程期间使用所述连续的气氛时,原料(例如,Co(OH)
焙烧过程的温度应为至少300℃并且至多800℃。如果温度低于300℃,则还原和氧化将不会发生。如果温度高于800℃,则钴氧化物可能被过度烧结。例如,焙烧温度可为约300℃至700℃、约300℃至600℃、约300℃至500℃或约350℃至450℃。在另外的示例中,焙烧温度可为约300℃、325℃、350℃、375℃、400℃、425℃、450℃、475℃、500℃、525℃、550℃、575℃或600℃。非氧化气氛的焙烧温度可与氧化气氛的焙烧温度相同。另选地,非氧化气氛的焙烧温度可不同于氧化气氛的焙烧温度。
焙烧时间应足够长,以还原和氧化引入加热炉中的至少绝大部分粉末。因此,焙烧时间将取决于诸如粉末量和加热炉的类型等因素。一般来讲,静态炉的焙烧时间为至少一小时且至多15小时。例如,焙烧时间可为约1小时至10小时、约1小时至8小时、约2小时至6小时、或约2小时至4小时。在另外的示例中,焙烧时间可为1小时、2小时、3小时、4小时、5小时、6小时、7小时、8小时、9小时、10小时、11小时、12小时、13小时、14小时或15小时。非氧化气氛中的焙烧时间可与氧化气氛中的焙烧时间相同。另选地,非氧化气氛中的焙烧时间可不同于氧化气氛中的焙烧时间。
通过焙烧工艺制备的钴氧化物(其中含钴原料首先在非氧化气氛中焙烧,然后在氧化气氛中焙烧)比通过具有单一类型气氛的焙烧工艺所制备的钴氧化物具有更高的机械强度。此外,所得的LCO化合物(即,使用通过所述两种气氛连续的焙烧工艺制备的钴氧化物前体生成的那些LCO化合物)具有优越的电化学特性。事实上,具有小于15μm的平均粒度但通过所述两种气氛连续的焙烧工艺制备的钴氧化物前体比具有类似尺寸和组成但通过单一气氛焙烧工艺制备的钴氧化物前体具有更高的机械强度。但是,尽管具有较高的机械强度,使用平均粒度小于15μm的钴氧化物前体制备的LCO化合物(无论制造方法如何)电化学特性和体积密度较差。
在操作上,可使用各种炉配置利用两种不同连续的气氛进行焙烧。例如,可使用两个分开的加热炉来实现两种气氛连续的焙烧。利用分开的加热炉,第一焙烧可在具有非氧化气氛的加热炉中按指定的温度和时间进行。第二焙烧可在具有氧化气氛的后续独立加热炉中按指定的温度和时间进行。另选地,也可使用单一加热炉,其中气氛从用于第一焙烧的非氧化气氛变为用于第二焙烧的氧化气氛。
在实施例中使用以下分析方法:
使用以
通过扫描电镜(SEM)技术分析正电极材料的形态。使用JEOL JSM7100F扫描电镜设备在9.6×10
其中代表性前体颗粒的表面积(A)和周长(P)通过使用熟知的基于ImageJ的软件获得(参考ImageJ用户指南的“设定测量”的第30.2至30.7节,https://imagej.nih.gov/ij/docs/guide/user-guide.pdf或https://imagej.nih.gov/ij/docs/guide/146-30.html)。
如上所述,圆度的计算意味着对以下内容的测量:
i)所述周长通过以下方式确定:a)确定颗粒的SEM图像的外边界,b)将所述外边界分解成基于单个分段的选择项,这些选择项中的每一者均具有单个周长,以及c)将所述单个周长的长度值相加以便获得颗粒的周长值;以及
ii)所述面积通过以下方式计算:将包含在由所述外边界限定的表面中的多个像素面积相加。
圆度为1.00意指代表粉末样品的颗粒具有球形形状。
圆度小于1.00意指代表粉末样品的颗粒具有非球形形状。
圆度大于0且小于1是指椭圆形形状。
通过使用压缩强度的测量来研究前体颗粒的强度。此外,在施加诸如50MPa等特定压力之前和之后测量粒度分布(PSD)的变化。
通过研究施加压力后PSD的变化来分析颗粒强度。首先,将前体粉末置于直径“d”为1.3cm的不锈钢粒料模头中。施加50MPa的单轴压力。然后用手指或研钵轻轻地拆开所得的粒料,以获得用于PSD测量(于下文E中描述)的散粉。
为了评估前体颗粒的压缩强度,使用微压缩测试仪(Shimadzu,MCT-W500-E)。选择十颗具有平均粒度的代表性颗粒。用连续增大的力按压每颗代表性颗粒直至其破碎。用于破碎各个颗粒所施加的力的最小值被认为是该颗粒的压缩强度(MPa)。
如下测量压制密度(PD):将3g的粉末填充到直径“d”为1.3cm的粒料模头中。向粒料施加207MPa的单轴压力并持续30秒。松弛负载后,测量压制粒料的厚度“t”。然后如下计算压制密度:
压制后,通过E)PSD测量进一步研究粉末。
在将粉末分散在含水介质中之后,使用具有Hydro MV湿分散附件的MalvernMastersizer 3000分析颗粒压制测试或PD测量之前和之后的粉末的粒度分布(PSD)。为了改善粉末在含水介质中的分散,施加足够的超声辐射和搅拌,并引入合适的表面活性剂。如本文所用,D10、D50和D90被定义为累积体积%分布的10%、50%和90%处的粒度。
在颗粒压制测试之前和之后测量D10。如下计算在50MPa下D10从压制前到压制后的变化:
在PD测量之前和之后测量D10。如下计算在207MPa下D10从压制前到压制后的变化:
在50MPa或207MPa的压力下压制后,D10的变化可用作对粉末的损坏进行定量以及因此对粉末的强度进行定量的标准。
通过电感耦合等离子体(ICP)方法使用Agillent ICP 720-ES测定粉末样品的组成。在锥形烧瓶中将2g粉末样品溶解于10mL高纯度盐酸中。该烧瓶用玻璃覆盖并在热板上加热以完全溶解前体。冷却至室温之后,将溶液移至100mL容量瓶中,使用蒸馏(DI)水冲洗该烧瓶3至4次。之后,用DI水填充容量瓶直至100mL刻度,然后完全均化。用5mL移液管取出5mL溶液,并转移到50mL容量瓶中以进行第2次稀释,此时在该容量瓶中填充10%盐酸直至50mL刻度,然后均化。最后,将此50mL溶液用于ICP测量。
通过元素Co和元素A相对于ICP分析中粉末样品的总重量的重量比来获得钴氧化物前体的式Co
为了制备正电极,通过高速均质器制备在溶剂(NMP,Mitsubishi)中包含按重量计90:5:5的电化学活性材料、导体(Super P,Timcal)、粘结剂(KF#9305,Kureha)的浆料。使用具有230μm间隙的刮涂刀涂覆器将均匀化浆液涂抹在铝箔的一面上。将经浆液涂覆的箔在120℃的烘箱中干燥,然后使用压延工具压制。然后再次在真空烘箱中干燥,以完全移除电极膜中的剩余溶剂。纽扣电池在充满氩气的手套箱中组装。隔膜(Celgard 2320)位于正电极和用作负电极的锂箔片之间。将含1M LiPF
本发明中的纽扣电池测试(其为常规“恒定截止电压”测试)遵循表1中所示的计划。
使用Toscat-3100计算机控制的恒电流循环站(来自Toyo)将每个电池在25℃下循环。纽扣电池测试程序在4.50-2.75V/Li金属窗口范围内使用185mA/g的1C电流定义。
CQ1(mAh/g)和DQ1(mAh/g)分别为第一次循环的充电和放电容量。
纽扣电池测试程序使用225mA/g的1C电流定义。该测试方法是在4.60-2.75V/Li金属窗口范围内评估0.2C时的循环寿命。表2示出了纽扣电池测试计划。
剩余容量(以%计的QR)如下计算:
DQ1(mAh/g)是第一循环的放电容量。DQ50(mAh/g)是第五十循环的放电容量。
本发明在以下实施例中进一步举例说明:
Co
然后将钴原料CEX1.1的一部分于400℃处在N
为了制备示例性LCO产品,将EX1和Li
研磨烧结的凝聚LCO化合物。因此,形成了Li
CEX2使用用于EX1的相同方法制备,不同的是当沉淀浆液的粒度达到7μm时停止沉淀。
LCO3使用与LCO1相同的程序制备,不同的是CEX2代替EX1用作钴前体。
CEX2和LCO3的分析结果也分别在表4和表5中示出。
CEX3为WO'165中公开的钴氧化物(Co
EX2使用用于EX1的相同方法制备,不同的是将CoCO
LCO4使用与LCO1相同的程序制备,不同的是EX2代替EX1用作钴前体。另外,将EX2转化成LCO4的烧结温度为940℃。
EX2和LCO4的分析结果也分别在表4和表5中示出。
如表4所示,非焙烧的Co
通过在N
根据EX1制造的LCO1具有最佳性能。具体地讲,LCO1体现了如表5所示的第五十次循环(QR)之后的高初始放电容量和最高剩余容量。图1还示出了LCO1的优越性能。图1为示出用作循环次数函数的LCO1和LCO2.1至LCO2.3的剩余容量的图表,其中x轴为循环次数,并且y轴为剩余容量。图1示出LCO1在多次循环之后具有高剩余容量。具体地讲,LCO1在40次循环之后具有最高的剩余容量。
EX1的高颗粒强度促成了LCO1的优越蓄电池性能。图2A提供了EX1和LCO1的PSD曲线的比较。可以看出,EX1和LCO1之间的PSD曲线差异非常小,表明EX1的高压缩强度使得在制备LCO1的过程期间发生的颗粒损坏非常小。另外,如表4所示,EX1具有0.89的圆度(这非常类似于产生EX1时焙烧的钴原料的圆度),表明即使在钴原料的焙烧过程之后仍保持球形颗粒。
前体颗粒的组成也可影响所得LCO材料的电化学特性。以下示例示出了这一点。
图2B提供了CEX1.1和LCO2.1的PSD曲线的比较。如图2B所示,尽管CEX1.1具有较高的压缩强度,但CEX1.1和LCO2.1的粒度分布存在较大的差异。另外,LCO2.1具有相对较差的电化学特性。
图3示出了在锂化过程中用于制造LCO1和LCO2.1的托盘的示意性模型。在锂化过程之后,从托盘的“a”部分至“e”部分获取LCO1和LCO2.1粉末的示例性样品,并且通过ICP分析来分析它们的Li/M'比率。
该样品的ICP分析结果在图4中示出。可以看出,LCO2.1样品的Li/M'比率在托盘中全程发生变化(即,不是恒定的)。取样颗粒表明,Li/M'比率基于整个托盘中的平均Li/M'比率而变化约15%。期望将整份粉末中Li/M'比率的百分比变化保持小于约5%,优选地小于约3%。如上所述,不均匀的Li分布是由锂化反应期间作为副产物产生的H
从表5中可以看出,由D50为7.9μm的钴氧化物制备的LCO3具有较差的电化学特性和低体积密度。
于940℃处使用无掺杂的Co
所有上述LCO1、LCO2.1至LCO2.3、LCO3和LCO4均包含具有R-3m晶体结构的颗粒。
本发明通过以下项目进一步限定:
1.一种用于制备正电极活性材料的钴氧化物前体粉末,其中前体组合物包含具有式Co
2.一种正极活性材料,该正极活性材料由根据项目1所述的钴氧化物前体粉末制备。
3.根据项目2所述的正极材料,该正极材料具有等于或大于3.7g/cm
4.根据项目2或3所述的正极活性材料,其中该材料具有至少192mAh/g的DQ1。
5.根据项目4所述的正极活性材料,该正极活性材料在五十次循环之后具有至少40%、优选地至少50%的剩余容量(QR%)。
6.根据项目5所述的正极活性材料,其中:y>0或其中:y>0,并且A包含Mn。
7.一种用于制备根据项目1所述的钴氧化物前体粉末的方法,该方法包括以下步骤:
-在干燥惰性气氛中,特别是在干燥氮基气氛中,将包含钴盐颗粒(如Co(OH)
-在干燥氧化气氛中,将包含中间体前体颗粒的组合物于至少300℃至不超过600℃的温度下加热2-5小时,以形成包含钴氧化物前体颗粒的粉末。
8.一种制备用于二次蓄电池的基于锂钴氧化物的活性材料粉末的方法,该方法包括以下步骤:
-提供根据项目1所述的前体粉末,
-提供含Li前体粉末并将所述含Li前体粉末与根据项目1所述的前体粉末混合,以及
-在5-20小时的时间段期间、于至少900℃且至多1200℃的温度下在含氧气氛中烧结所述混合物,从而获得所述基于锂钴氧化物的活性材料粉末。
9.根据项目8所述的方法,其中根据项目1所述的前体粉末与所述含Li前体粉末以Li/(Co+A)>1.00的摩尔比混合。
10.一种制备根据项目2至6中任一项所述的基于锂钴氧化物的活性材料粉末的方法,该方法包括以下步骤:
-提供根据项目1所述的前体粉末,
-提供含Li前体粉末并将所述含Li前体粉末与根据项目1所述的前体粉末混合以获得混合物,以及
-在5-20小时的时间段期间、于至少900℃且至多1200℃的温度下在含氧气氛中烧结所述混合物,从而获得所述基于锂钴氧化物的活性材料粉末。
11.根据项目10所述的方法,其中根据项目1所述的前体粉末与所述含Li前体粉末以Li/(Co+A)>1.00的摩尔比混合。
- 用作可再充电锂离子蓄电池的正电极材料的前体的钴氧化物
- 用于可再充电锂离子蓄电池的正电极材料的前体