DNA-细胞缀合物
文献发布时间:2023-06-19 12:04:09
本申请是申请日为2010年4月8日、申请号为201710671296.3、发明名称为“DNA-细胞缀合物”的中国发明专利申请的分案申请。
相关申请的交叉引用
本专利申请主张2009年4月8日提交的美国专利申请No.61/167748和2009年9月16日提交的美国专利申请No.61/243123的优先权,以上专利申请出于所有目的以其整体并入本文。
有关在联邦政府资助的研究或开发下完成的发明权利的声明
本发明是在美国能源部所签订的并且由能源部和科学局、基础能源科学局的纳米尺度科学、工程和技术(NSET)项目所支持的合同No.DE-AC02-05CH11231、国家卫生研究院授予的资助号No.HG003329和R01 GM072700以及国家卫生研究院分子生物物理学培训资助计划No.T32GM08295的政府支持下进行的。政府对本发明具有一定权利。
对通过光盘提交的“序列表”、表格或计算机程序列表附录的参考
不适用
发明背景
通常,已经在基于细胞的阵列中使用肽来捕获细胞用于研究。表面印制了设计用于结合细胞表面上整合素的“RGD”肽,但是该系统不能用于不具有整合素的细胞如非粘附细胞(例如白细胞、淋巴细胞),并且不允许在同一平台上用不同细胞类型共同进行受控的限定的图案化。由于整合素是参与控制分化的受体,并且整合素结合后的活性与分化有关,因此RGD系统具有引起细胞分化并因此在进行分析之前改变细胞的巨大劣势。(参见Du,X.P.等人,Cell 1991,65,409-416;Xiong,J.P.等人,Science 2002,296,151-155。)
使用蛋白质-蛋白质连接系统证明了间接非共价连接,其中DNA通过抗体-配体相互作用以非共价键间接地与细胞连接(Bailey RC等人,J Am Chem Soc.2007Feb 21;129(7):1959-67)。将针对细胞上靶配体特异性的抗体与DNA缀合。抗体和配体之间的非共价键是基于氢键作用的,即典型的蛋白质-蛋白质相互作用。将细胞表面配体特异性抗体上的单链DNA(ssDNA)寡聚物与具有那些配体的细胞相连,然后将所述细胞锚定到具有ssDNA互补寡聚物的表面上,所述ssDNA互补寡聚物与所述细胞上的配对链结合以捕获所述细胞。
参见Chandra,R.A.等人,Angew.Chem.-Int.Edit.2006,45,896-901使用的代谢寡糖工程首先显示了合成单链DNA(ssDNA)链与活细胞表面的间接共价连接。这种间接共价是如下所述进行的:通过在引入DNA前用过乙酰化的N-叠氮基乙酰甘露糖胺(Ac4ManNAz)处理细胞(花费3天)后所获得的细胞表面唾液酸中所引入的特定化学柄(chemical handle)(叠氮化物)进行的。然后,将膦-ssDNA缀合物共价连接到叠氮化物柄上从而在叠氮基糖和DNA之间形成了酰胺键(施陶丁格连接反应,E.Saxon等人,Science 2000,287)。通过合成叠氮基糖的代谢在细胞表面糖缀合物内设置的叠氮化物可以与生物素化的三芳基膦反应以产生多种稳定的细胞表面加成物。然而,共价代谢方法花费数天时间来制备用于DNA连接的细胞,并且改变细胞表面的糖对细胞有代谢上的影响,其在有机会对所述细胞进行分析之前使其改变。它还限于在表面上具有唾液酸的特定哺乳动物细胞,因此不能用于细菌、植物细胞、真菌或多种其他动物细胞。
通过抗体和细胞表面上配体的非共价连接还将活化细胞并因此在可以开始分析前干扰细胞,另外,与共价连接相比,非共价连接倾向于较弱并且是“可逆的”。另外,抗体机制需要在细胞上普遍存在配体,并且需要设计对配体具有特异性的抗体从而将足够的DNA固定在细胞表面上。
已制备了通过细胞表面上的配体与抗体之间相互作用的非共价连接,其中所述抗体带有结合蛋白的DNA链(Bailey RC等人,J Am Chem Soc.2007Feb 21;129(7):1959-67)。这两种方法均具有使得它们所要捕获用以研究的细胞活化的直接缺点,并因此在可以进行分析之前将目标事物变得不同。克服这些细胞表面上DNA连接早期系统的缺点可改变此所述新兴领域并提供先前所不可能的有价值工具和操作。
发明内容
在一个实施方案中,本发明提供了具有细胞的组合物,其中所述细胞具有包含天然官能团的表面,并且其中所述细胞没有细胞壁。所述组合物还包含核酸部分,其中所述核酸部分共价连接到所述天然官能团上。
在另一个实施方案中,本发明提供了通过将细胞与活化的核酸部分相接触来制备所述细胞和核酸部分的缀合物的方法,其中所述细胞具有包含天然官能团的表面,并且其中所述细胞没有细胞壁,从而使得所述核酸部分共价连接到所述天然官能团上。
在另一个实施方案中,本发明提供了具有细胞和核酸部分的组合物,其中所述细胞具有细胞壁,以及其中所述核酸部分共价连接到所述细胞。
在另一个实施方案中,本发明提供了通过将细胞与活化的核酸部分接触来制备所述细胞和核酸部分的缀合物的方法,其中所述细胞包含细胞壁,从而使得所述核酸部分与所述细胞连接。
在另一个实施方案中,本发明提供了包含细胞和基底表面的装置,其中所述细胞具有共价连接到第一核酸部分的天然官能团的细胞表面,所述基底表面具有与所述第一核酸部分互补的第二核酸部分,使得所述细胞通过形成第一和第二核酸部分的核酸双链体从而与所述基底表面结合。
附图说明
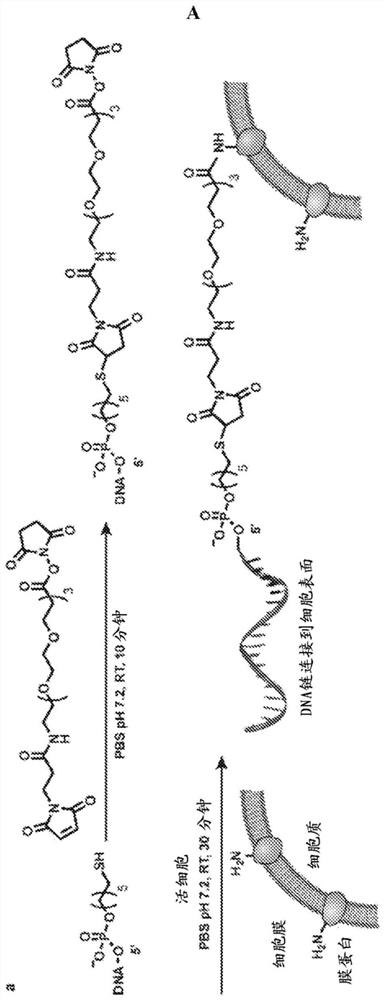
图1.ssDNA与细胞表面的共价连接。(a)首先,在室温下将巯基化单链DNA与PBS中的NHS-PEG-马来酰亚胺反应以形成NHS-DNA缀合物。然后,在室温下将该溶液与PBS中的活细胞悬液一起孵育30分钟。连接DNA链之后,将细胞放回到培养基中。(b,c)将Jurkat细胞暴露于(a)中所述的不同浓度的NHS-DNA溶液。然后,加入荧光互补链,并使用流式细胞术对细胞修饰水平定量。在每个细胞上可以装载多达120000条DNA链。(d)使表面上细胞图案化的方法的示意图。
图2.以DNA序列特异性的方式固定细胞。(a)具有与DNA微阵列上的互补(序列M2)点(点尺寸=60μm)相结合的DNA序列C2的细胞。具有非互补序列M1的相邻点保持为空。(b)将同一微阵列基底分别暴露于具有序列C2和C1的Jurkat和MDA细胞的混合悬液。用Cell-Tracker Green对Jurkat细胞染色,并且用Cell-tracker Blue对MDA细胞染色。(c)MCF-7和(d)MDA细胞还以快速、稳定并且序列特异的方式与DNA包被的表面结合。在DNA包被和未包被区域之间观察到了清楚的界限。孵育2小时后,显示了相差图像。(e)MCF-7和(f)MDA细胞在36小时后铺展并增殖,但它们仍被约束在DNA印制区域内。
图3.原代细胞的直接DNA修饰和捕获。(a)人红细胞以与Jurkat细胞相同的方式结合在DNA点上,并且它们在刚结合后看上去在形态上是相同的。台盼蓝染色表明膜保持完整。(b)DNA包被的小鼠CD4+辅助T细胞与包被了互补DNA的点结合。暴露3分钟后,在载玻片的印制和未印制区域之间可以观察到清楚的边界。(c)通过光刻法和微制造(microfabrication)制备了微尺度的DNA图案。与荧光素缀合的ssDNA链在基底上形成图案以允许观察。(d)在相同DNA图案上捕获了小鼠原代T细胞。(e)DNA固定的T细胞和游离T细胞的IL-2生产,如通过ELISA所确定的。ConA=刀豆蛋白A。PMA=醋酸佛波醇肉豆蔻酸酯。CSA=环孢菌素A。
图4.原代成肌细胞的捕获和分化。(a,b)载玻片上的DNA图案指明了结合细胞的区域。(c)成肌细胞在刚捕获时(如所示)或在生长培养基中保持1天后未显示出分化迹象。(d)一旦加入分化培养基,则形成了肌管。在转变进行5天后照相。(e)在分化培养基中孵育6天后,显示圆形图案的成肌细胞形成了与边缘对齐的圆弧形肌管。(f)6天后,矩形布置中的肌细胞形成了与图案长轴对齐的肌管。对于线形图案,大部分肌细胞(g)排列在20°的图案边界角内并且(h)存在于边缘之间。
图5.DNA修饰反应的基质辅助激光解吸电离/飞行时间(MALDI-TOF)质谱。发现模式胺化合物与NHS酯反应,从而证实了细胞表面上酰胺的形成。
图6.经NHS-DNA修饰的Jurkat细胞的存活。A)将DNA包被的细胞(各20个碱基)溶液与互补链混合。在多个时间点,对细胞总数目视计数(蓝色曲线)。对照样品(桔黄色曲线)由未加入DNA的情况下生长的未修饰Jurkat细胞组成。B)为了评价连接后的存活,将经DNA修饰的细胞固定在具有互补链的载玻片上。在固定24h和48h后,将细胞与膜联蛋白V-FITC(绿线条)和PI(红线条)的溶液一起孵育。在1h内,使用荧光显微镜评价细胞。游离细胞是缺少表面DNA并且不与载物片结合的对照样品。
图7.在2、12、24、36小时后,对固定的MCF-7和MDA细胞成像。
图8.将成肌细胞在生长培养基中孵育3天。A)将成肌细胞接种到包被胶原的培养皿上。B)成肌细胞与NHS-DNA反应,并以序列特异性方式与表面结合。
图9.在2、4、6、8、10、12、14天后将在融合培养基中孵育的成肌细胞接种到包被胶原的培养皿中或与NHS-DNA反应并以序列特异性方式与表面结合。
图10.限定图案上的肌管排列分析。测量了肌管和最近的边缘之间的距离(红线)和角度。所有距离测量均是从管中点开始的。用绿线表示图案边缘之间的全长距离。
图11.用于单细胞监测的双功能微电极阵列的制造。(A)使用光刻法和剥离法(liftoff)在玻璃晶片上使金电极图案化。然后,将7μm的聚氯代对二甲苯(Parylene-c)绝缘层沉积到电极上,并用100nm的蒸发铝层覆盖。(B)在铝层上将光刻胶图案化,然后对铝层进行蚀刻并将其用作聚对二甲苯(Parylene)绝缘体的蚀刻掩模。(C)将氧化铱传感器层沉积到电极表面上,然后用醛基硅烷处理以用于胺修饰的捕获DNA连接。(D)最后,将铝层溶解于强碱中,从而在传感器表面上仅留下捕获DNA。引入具有与表面结合的互补链的细胞,其直接并特异性地捕获到传感器上。(E)用具有与细胞表面上伯胺结合的末端N-羟基琥珀酰亚胺基(NHS)酯官能团的单链DNA(5'-CCCTAGAGTGAGTCGTATGA-3')处理细胞。该DNA条形码标记使装置中用于DNA靶向捕获的细胞官能化。(F)微流体装置简图。用PDMS通道包封电极,从而形成了微流体装置。
图12.双功能微电极阵列的校准数据。(A)使用标准pH4、5和7的缓冲液,对一个DNA修饰的氧化铱传感器所进行的典型校准记录。相对于Ag/AgCl参比电极测量了电压。(B)电压相对于pH标准品测量的曲线,其斜率为-68.5mV/pH单位,R
图13.双功能微电极阵列上的细胞捕获。个别非粘附Jurkat细胞的荧光显微照片,所述细胞具有与传感器电极上互补链结合的表面结合DNA条形码。用白色标出电极区域。标尺=40μm。插图:电极上单个Jurkat细胞的放大视图,其具有额外的侧面照明以显示电极区域。
图14.用双功能微电极阵列测量的单细胞酸化。(A)在已知同类样品中测量的单个Jurkat和原代T细胞酸化的代表性综合数据。(B)从混合物中捕获的单个Jurkat和原代T细胞,并且在阵列上同时对它们监测了10分钟的时间跨度。(C)在10分钟内,已知类型样品中个体细胞酸化的柱状图。在低缓冲培养基中,观察到Jurkat细胞具有比原代T细胞显著更高的酸化率(P<0.0002)。
图15.通过双功能微电极阵列测量的单细胞刺激。Jurkat细胞在前13分钟内表现出正常的基线酸化,然后将125μL 10μM鱼藤酮在低缓冲培养基中的溶液加入到通道出口储库中,在此处它在几秒钟内扩散到通道内。鱼藤酮抑制线粒体电子传递链,从而导致乳酸分泌速率提高,并因此导致了更高的酸化速率。
图16.生物分子与悬臂和细胞表面的共价连接。a)使用氧等离子体使表面氧化后,使用化学气相沉积(chemical vapor deposition,CVD)将醛官能团引入到氮化硅悬臂上。b)在潮湿室中,将含有硼氢化钠的抗CD3 IgG或ConA的溶液引入到醛包被的悬臂表面上(IgG=免疫球蛋白G)。通过在100℃下将悬臂浸入胺官能化的ssDNA溶液中30分钟并随后暴露于硼氢化钠溶液来实现DNA修饰。c)通过用过乙酰化的N-叠氮乙酰基甘露糖胺(Ac
图17.基于生物分子的粘附方法的比较。a)首先,在粘附分子存在下确定整体细胞生长速率。将Jurkat细胞悬液与ConA或抗CD3 IgG混合,将DNA包被细胞的溶液与互补DNA链混合。在多个时间点,对细胞总数计数。在无任何粘附分子的情况下,培养对照样品。b)为了评价细胞捕获效率,将20μM FITC-标记的ssDNA、20μM FITC-标记的ConA和6μM FITC-标记的抗CD3 IgG溶液施加到醛包被的载玻片上,并通过还原性胺化连接生物分子。然后,将含有1×10
图18.脱粘作用力(de-adhesion)的AFM测量。A)用灰度显示了六个单细胞样品的踪迹,用黑色显示平均踪迹。在零距离处,细胞与悬臂完全接触,悬臂施加正作用力。随着距离增加,悬臂从载玻片表面上拉离,造成细胞-悬臂连接断裂并导致产生了零作用力和无接触区。将脱粘作用力计算为最小弯曲和水平无接触区之间的差值。B)在不同的收缩速率(15.7和8.2μm/s)和对DNA、ConA和抗体系统的接触力(400和200pN)下测量了粘附力。通过对不止4种不同的细胞测量了六个脱粘事件获得了数据。误差线代表一个标准差。
图19.活细胞的蘸水笔(dip-pen)图案化。a)通过将较短的DNA链(13个碱基)连接到悬臂并且将较长的链(20个碱基)连接到载玻片上,可以通过AFM仪传输单个活细胞并将其直接印制在载玻片上的所需位置。b-d)该方法显示了形成细胞单个图案的步骤。
图20.单细胞基因沉默测定概述。培养Jurkat细胞并进行表面标记,通过形成DNA双链体在靶标垫上捕获单个细胞,并且产生了RT-PCR表达谱。(A)与对照18S rRNA相比,正常生长条件下的细胞显示出均匀的GAPDH高表达(绿色细胞)。(B)用靶向GAPDH的siRNA处理的细胞显示出不同水平的mRNA抑制。
图21.微流体装置布局图。示意图显示了用于单细胞基因表达谱分析的半个装置(4个完整系统中的2个)。4层玻璃-PDMS-玻璃-玻璃微装置含有4个单独的区域。顶部的第一个区域为3阀泵。反应器区由位于200nL反应室中心的光刻限定的细胞捕获金垫以及用于热循环的RTD和微制造加热器组成。亲和捕获区包含容留室和亲和捕获腔室(黄色)。最后,在CE分离通道(红色)上分析了热释放的扩增子。每个装置含有4个独立可寻址系统,其使得能够平行地进行4个单细胞的分析。将所有通道蚀刻至20μm深。
图22.在集成基因表达微装置中实施的生物化学步骤的示意图。(上图)分析在<75分钟内完成。(下图)(A)单细胞基因表达微系统的操作说明。(B)首先,将在细胞膜上用20个碱基的寡核苷酸官能化的细胞流入反应器中。(C)当细胞外部的ssDNA与固定在金垫上的互补捕获链结合时,在尺寸限制的25×25μm
图23.单细胞水平的基因表达和沉默。(A)个体Jurkat细胞的代表性基因表达电泳图。具有靶向GAPDH(200bp)和18S rRNA(247bp)的引物的单个野生型细胞在160s和185s分别产生了2个强烈的迁移峰。用靶向GAPDH mRNA的siRNA电穿孔的单个细胞仅显示出对应18S rRNA的单个峰。(B)相对于正常未处理的细胞,用GAPDH siRNA处理的Jurkat细胞的GAPDH基因表达。将GAPDH表达相对于对照18S rRNA进行归一化用以比较。8个个体细胞的实验显示GAPDH mRNA水平为正常未处理的Jurkat细胞的0、5、50、1、48、0、5和0%。然而,50个细胞的代表性整体测量显示GAPDH表达为21±4%。当在垫上未捕获到细胞时,没有扩增。类似地,无逆转录酶的PCR对照显示无扩增。(C)siRNA处理的细胞的事件数的柱状图显示有2个不同的细胞群体,它们的表达水平与群体平均值相差很大。
图24.显示了用于靶向递送的衣壳的双面修饰。对于内表面修饰,MS2衣壳蛋白的N87C突变使得能够进行位点特异的烷基化。可以在这些位置上安装多达180个装载分子。对于外表面修饰,首先用苯二胺基团修饰适体(aptamer)。衣壳上的T19paF突变使得能够通过NaIO
图25.DNA与MS2外表面连接的分析。(a)通过SDS-PAGE,随后用考马斯染色分析了MS2-DNA缀合物。泳道1-7与链A反应。泳道8显示了与链B的反应。(b)凝胶迁移测定确认了在与MS2缀合后,DNA的碱基配对能力。泳道10包括链A的互补序列,其具有额外的20个腺嘌呤碱基以提高电泳迁移,而泳道9不具有加入的额外DNA。透射电子显微镜照片(c)和动态光散射分析(d)显示了DNA缀合后完整的衣壳。另外,DLS显示与链A结合后直径显著增加,并且加入与链A互补的20碱基序列(无额外的20个碱基突出),直径进一步增加。TEM图中的比例尺代表100nm。
图26.适体标记衣壳的细胞靶向和摄取。(a)使用流式细胞术确认了细胞靶向。仅有用链B修饰的MS2衣壳与Jurkat细胞(蓝色)结合。外部未修饰的衣壳和用链C修饰的衣壳(分别为绿色和黄色)显示B标记衣壳与LDL标记内体(endosome)(b)共定位,但是不与转铁蛋白标记内体(c)共定位。比例尺线表示3μm。
图27显示了内部装载物的安装细胞靶向适体与病毒外部的连接。示意图显示了涉及病毒的基本过程:用寡核苷酸适体在碳水化合物处包被病毒,所述病毒含有装载物(多达180个分子);通过适体对靶标活细胞表面上细胞表面蛋白的特异性,适体与受体蛋白结合并将其内容物内化或递送到该细胞中。
图28显示了使用本发明方法图案化的细胞的图片以及对生物在协同太阳能H2燃料电池(芯片上)中作用的简短解释。图28A显示了集胞藻(Synechocystis)PCC6803(蓝细菌)、莱茵衣藻(C.reinhardtii)(藻)、深红红螺菌(R.rubrum),图28B显示了棕色固氮菌(A.vinelandii)。将细胞在大的ssDNA点上图案化,这是图中仅显示图案部分曲率的原因。
图29显示了大肠杆菌(E.coli)图案的图片(A)显示了图案化的用FITC DIC叠加标记的大肠杆菌(E.coli),20×;(B)和(C)显示了图案化大肠杆菌(E.coli)(未荧光标记)和集胞藻(Synechocystis)(用FITC标记)的微阵列图案,20×(上部分)和10×(下部分)。存在背景荧光,但通过它们的纹理可指示细胞。
图30显示了用于燃料电池装置中的玻璃基底上集胞藻(Synechocystis)PCC6803(蓝细菌)、棕色固氮菌(A.vinelandii)和深红红螺菌(R.rubrum)图案化层的示意图。
图31显示了通过本发明与细胞表面上赖氨酸天然官能团的反应所制备的哺乳动物细胞的DNA-细胞缀合物,以及将所述缀合物固定在互补DNA链修饰的显微镜用载玻片上,证明形成了DNA-细胞缀合物。
图32显示了在用互补DNA链修饰的显微镜用载玻片上固定了通过本发明所制备的非哺乳动物细胞的DNA-细胞缀合物,证明形成了DNA-细胞缀合物。
图33显示了通过本发明与细胞表面上的天冬氨酸或谷氨酸天然官能团的反应所制备的哺乳动物细胞的DNA-细胞缀合物,以及将所述缀合物固定在互补DNA链修饰的显微镜用载玻片上,证明形成了DNA-细胞缀合物。
发明详述
I.概要
本发明提供了第一次在不需要代谢工程的情况下通过细胞表面上的天然官能团将DNA共价连接到细胞上从而制备了细胞-DNA缀合物的实例。对于哺乳动物细胞以及其他无细胞壁的细胞,将DNA直接共价连接到所述细胞表面上的氨基酸,如赖氨酸胺。对于植物细胞以及其他具有细胞壁的细胞,首先修饰细胞表面上的糖,如通过氧化形成醛或酮,然后用DNA修饰。
先前形成DNA-细胞缀合物的方法需要使用代谢工程,即,用适当修饰的糖(如叠氮化物官能化的糖)喂养细胞,该糖被细胞所代谢并在细胞表面上表达。然后,将DNA缀合到叠氮化物修饰的糖。该方法具有一些缺点,限制了其应用的用途和范围。例如,代谢工程需要几天时间来提供在细胞表面上具有足够量叠氮化物官能化糖的细胞。可以修饰的细胞类型也是有限的,排除了具有重要诊断应用的原代细胞。最后,代谢工程必然改变细胞的结构和特性。
本发明通过使用细胞表面上已存在的天然官能团克服了叠氮化物代谢工程方法中固有的困难。使用活化DNA序列,如NHS酯所修饰的DNA,可以将DNA共价连接到无细胞壁细胞的细胞表面上赖氨酸基团的胺上。该方法最多花费几小时的时间。该方法的简单性使得能够对原代细胞以及干细胞进行修饰。对于具有细胞壁的细胞,首先修饰细胞表面上的天然糖,其通过(例如)氧化形成随后将与活化DNA序列反应(如与氨基氧基团反应)的醛和酮来进行。本发明的DNA-细胞缀合物和方法代表了细胞检测和测定方法的巨大飞跃。
在表面上使细胞图案化的能力为多种研究和应用提供了新的平台,例如,细胞生物学研究、干细胞分化的控制和新组织的改造。通常,通过用设计与细胞表面上的整合素结合的“RGD”肽印制目的表面从而形成了基于细胞的阵列。尽管该方法已广泛用于多种细胞类型的固定化,但是它不能用于捕获非粘附细胞(如白细胞)或将多种细胞类型与独特的阵列特征结合。由于它与参与控制这些过程的表面受体结合,因此它还可以导致细胞分化或行为发生不希望的变化。
为了克服这些限制,先前一些发明人在Chandra,R.A.;Douglas,E.S.;Mathies,R.A.;Bertozzi,C.R.;Francis,M.B.Angew.Chem.-Int.Edit.2006,45,896-901中已报道了通过共价连接到其质膜上的合成DNA链与印制了互补序列的表面的杂交来捕获活细胞,所述参考文献作为参考并入本文。除了能够使多种细胞类型在单个基底上图案化之外,该方法提供了基底的重复使用和可调节性的重要优势。最重要地,除了粘附细胞外,该方法已用于捕获非粘附细胞,并且由于通过这种不依赖于受体的方法进行固定化,已表明细胞在行为方面经历了最小的变化。在先前的报道中,还显示了该方法用于形成复杂细胞图案。
先前研究中所使用的DNA链是通过两步方法安装到细胞表面聚糖中的。首先,用含叠氮化物的甘露糖衍生物喂养细胞1-3天。随后,该糖被代谢并掺入到含有唾液酸的细胞表面聚糖中。然后,使用施陶丁格连接将DNA靶向到叠氮化物官能团。尽管有效,但是由于需要暴露几天来安装足够数目的叠氮基团,因此该方案最适合于培养的哺乳动物细胞系。
为了扩展基于DNA的粘附方法的通用性,本发明提供了将核酸直接安装到几乎任何细胞表面上的改进方法。下面参见图1,在一个实施方案中,首先将活化或官能化的单链核酸在缓冲溶液中与化学接头反应以形成核酸-接头缀合物。将细胞或细胞表面暴露于缓冲溶液一段指定的时间以允许反应进行从而将所述核酸通过化学接头连接到细胞表面上。在将核酸与细胞相连接后,使细胞返回培养基。如图1a中所述和定量的,可以使用不同浓度的核酸。在一个实施例中,将寡核苷酸在中性缓冲溶液中与化学接头反应以将寡核苷酸与化学接头相缀合,然后将所述细胞与含有寡核苷酸-接头缀合物的缓冲溶液一起孵育30分钟以允许修饰寡核苷酸-接头缀合物并将其连接到细胞表面上。
通常,通过在接头和细胞表面上的氨基酸之间形成共价键来进行细胞修饰。在一些实施方案中,细胞表面可以是任何来源的活细胞的细胞膜,其包括动物、植物、藻类或细菌细胞。在一些实施方案中,共价键为酰胺键或酯键。在一些实施方案中,寡核苷酸是单链的。在另一些实施方案中,所述氨基酸选自赖氨酸、半胱氨酸、酪氨酸、丝氨酸、天冬氨酸、谷氨酸和色氨酸。
在具有细胞壁的细胞例如植物细胞中,所述的键可以是腙、肟或胺等,其中所述连接通过高碘酸盐氧化然后形成腙、肟或胺而发生。在一个实施方案中,细胞上的碳水化合物经历氧化产生醛官能团,然后所述的醛与寡核苷酸-接头缀合物反应以用于植物细胞表面的直接细胞修饰。
在一些实施方案中,可以在不到1小时内进行该过程,其使用任何目的寡核苷酸序列产生同等水平的细胞表面官能化。本发明方法可以适用于多种实施方案,其包括捕获单个细胞以用于RT-PCR分析、活细胞与固体基底的连接以用于作用力测量或细胞图案化技术。在实施例中,我们证明了这种新标记方法用于捕获红细胞、原代T细胞和成肌细胞的用途,所述细胞全都是使用其他方法难以图案化的细胞类型。这种新技术极大地扩展了基于DNA的粘附策略的范围并且它足够简单从而能够在非专业有机合成实验室中使用。
因此,在一个实施方案中,本发明是组合物,其包含具有直接共价连接的寡核苷酸的细胞膜。所述细胞膜可以是整个完整细胞,从而所述组合物包含具有与其直接共价连接的寡核苷酸的整个细胞。可以将多个寡核苷酸连接到单个细胞上。所述细胞可以是活细胞。所述细胞可以是任何细胞,如真核细胞,其包括动物细胞或非动物细胞。“直接”修饰细胞或细胞膜表示在连接寡核苷酸之前细胞膜(细胞表面、细胞外部)是未修饰或改变的。具体地,由于连接是在细胞表面上的成分上进行的,因此直接表示寡核苷酸所连接的成分在与寡核苷酸共价连接前是未修饰的。上述方法在连接寡核苷酸之前均首先修饰细胞表面上的部分。
共价连接表示在彼此连接的两个分子之间形成新的共价键。所述的键通常是酰胺键或酯键,但是也可以是在寡核苷酸和细胞表面上的部分(如蛋白质、氨基酸或碳水化合物,或其他细胞表面实体)之间起到连接目的的任何键。
一个实施方案利于在快速且有效的方法中用NHS-DNA缀合物直接修饰细胞表面,从而使得几乎任何哺乳动物细胞能够在1小时内在具有互补DNA的表面上图案化。本发明所述的具体技术证明了使用一般与先前的整合素靶向技术不相容的几种细胞类型的能力,其包括红细胞、原代T细胞和成肌细胞。与先前报道的基于抗体和凝集素的方法相反,该固定化过程不活化原代T细胞。在这些研究中,成肌细胞高效地图案化并且在与表面连接后保持未分化。一旦更换到分化培养基中,在图案区域中心形成边缘对齐程度优良的肌管。这种新方案的可用性极大地扩展了基于DNA的连接策略用于人工组织产生和将活细胞掺入装置环境的适用性。
II.定义
“细胞”是指生命的基本功能单元,其包括原核和真核细胞。细胞的特征在于具有核或核心的内部以及细胞膜(细胞表面)。细胞还可以具有细胞壁。无细胞壁的细胞包括真核细胞、哺乳动物细胞和干细胞。具有细胞壁的细胞包括原核细胞和植物细胞。其他细胞在本发明中是有用的。
“天然官能团”是指对于细胞表面来说是天然的官能团,如氨基酸和糖,并且其与核酸部分反应形成本发明的缀合物。示例性氨基酸包括赖氨酸、半胱氨酸、酪氨酸、苏氨酸、丝氨酸、天冬氨酸、谷氨酸和色氨酸。其他氨基酸是有用的,如以下所述的那些。糖对于细胞表面也是天然的,其包括甘露糖、半乳糖和唾液酸以及如下所述的那些。天然官能团可以未修饰形式与核酸部分反应或者可以经修饰从而使它们的反应性更强。
术语“氨基酸”是指天然和合成氨基酸以及以与天然氨基酸类似的方式起作用的氨基酸类似物和氨基酸模拟物。天然氨基酸是遗传密码所编码的那些氨基酸以及随后经修饰的那些氨基酸,例如,羟脯氨酸、γ-羧基谷氨酸和O-磷酸丝氨酸。
“氨基酸类似物”是指具有与天然氨基酸相同的基本化学结构的化合物,即与氢、羧基、氨基和R基结合的α碳,例如高丝氨酸、正亮氨酸、亚砜甲硫氨酸、甲硫氨酸甲基锍盐。这些类似物具有经修饰的R基(例如正亮氨酸)或经修饰的肽主链,但保留了与天然氨基酸相同的基本化学结构。
“非天然氨基酸”不是遗传密码所编码的,并且可以但不一定具有与天然氨基酸相同的基本结构。非天然氨基酸包括(但不限于)铃兰氨酸、2-氨基己二酸、3-氨基己二酸、β-丙氨酸、氨基丙酸、2-氨基丁酸、4-氨基丁酸、6-氨基己酸、2-氨基庚酸、2-氨基异丁酸、3-氨基异丁酸、2-氨基庚二酸、叔丁基甘氨酸、2,4-二氨基异丁酸、锁链素、2,2'-二氨基庚二酸、2,3-二氨基丙酸、N-乙基甘氨酸、N-乙基天冬酰胺、高脯氨酸、羟基赖氨酸、别羟基赖氨酸、3-羟基脯氨酸、4-羟基脯氨酸、异锁链素、别异亮氨酸、N-甲基丙氨酸、N-甲基甘氨酸、N-甲基异亮氨酸、N-甲基戊基甘氨酸、N-甲基缬氨酸、萘基丙氨酸、正缬氨酸、鸟氨酸、戊基甘氨酸、哌可酸、硫脯氨酸、氨基苯丙氨酸、羟基酪氨酸和氨基酪氨酸。
“氨基酸模拟物”是指具有与氨基酸的一般化学结构不同的结构但是以与天然氨基酸类似的方式起作用的化合物。
在本文中,可以用一般已知的三字母符号或用IUPAC-IUB生物化学术语委员会推荐的单字母符号表示氨基酸。同样地,可以用一般接受的单字母编码表示核苷酸。
“保守性修饰的变体”适用于氨基酸和核酸序列。对于特定核酸序列,“保守性修饰的变体”是指编码相同或基本相同的氨基酸序列的核酸,或者在核酸不编码氨基酸序列时,是指基本相同的序列。由于遗传密码的简并性,大量功能相同的核酸编码任何给定的蛋白质。例如,密码子GCA、GCC、GCG和GCU均编码氨基酸丙氨酸。因此,在用密码子表示丙氨酸的每个位置上,可以在不改变所编码的多肽的情况下将密码子改变为任何相应的所述密码子。这些核酸变化是“沉默变化”,它们是保守性修饰的变化的一种。本发明中编码多肽的每一个核酸序列还说明了核酸的每一种可能的沉默变化。技术人员将承认可以改变核酸中的每个密码子(除通常作为甲硫氨酸的唯一密码子的AUG和通常作为色氨酸的唯一密码子的TGG外)以获得功能上相同的分子。因此,在每条所述序列中隐含了编码多肽的核酸的每一种沉默变化。
至于氨基酸序列,技术人员将了解造成编码序列中单个氨基酸或少部分氨基酸改变、添加或缺失的核酸、肽、多肽或蛋白质序列的个别取代、缺失或添加是“保守性修饰变体”,其中所述改变导致用化学上类似的氨基酸(即疏水性、亲水性、带正电荷、中性、带负电荷)取代氨基酸。示例性疏水性氨基酸包括缬氨酸、亮氨酸、异亮氨酸、甲硫氨酸、苯丙氨酸和色氨酸。示例性芳香族氨基酸包括苯丙氨酸、酪氨酸和色氨酸。示例性脂肪族氨基酸包括丝氨酸和苏氨酸。示例性碱性氨基酸包括赖氨酸、精氨酸和组氨酸。示例性具有羧酸侧链的氨基酸包括天冬氨酸和谷氨酸。示例性具有羧酰胺侧链的氨基酸包括天冬酰胺和谷氨酰胺。在本领域中,提供功能相似的氨基酸的保守性取代的列表是公知的。这些保守性修饰的变体是本发明的多态性变体、种间同系物和等位基因的补充并且不把它们排除在外。
下列八组各自包含彼此作为保守性取代的氨基酸:
1)丙氨酸(A)、甘氨酸(G);
2)天冬氨酸(D)、谷氨酸(E);
3)天冬酰胺(N)、谷氨酰胺(Q);
4)精氨酸(R)、赖氨酸(K);
5)异亮氨酸(I)、亮氨酸(L)、甲硫氨酸(M)、缬氨酸(V);
6)苯丙氨酸(F)、酪氨酸(Y)、色氨酸(W);
7)丝氨酸(S)、苏氨酸(T);和
8)半胱氨酸(C)、甲硫氨酸(M)
(参见,例如,Creighton,Proteins(1984))。
“糖”是指糖类,如单糖、二糖、寡糖或多糖。单糖包括(但不限于)葡萄糖、核糖、果糖、唾液酸、甘露糖和半乳糖。二糖包括(但不限于)蔗糖和乳糖。多糖包括(但不限于)纤维素、半纤维素和木质纤维素或淀粉。其他糖类在本发明中是有用的。
如本文中所使用的,术语“接触”是指使至少两种不同的物质相接触从而使它们可发生反应的过程。然而,应理解所得的反应产物可以直接由所加入的试剂之间的反应产生或者由来自一种或多种所加入的试剂的中间体产生,所述中间体可在反应混合物中产生。
“核酸部分”是指含有多个核苷酸或核酸的基团。示例性核酸部分包括(但不限于)寡核苷酸、脱氧核糖核酸(DNA)、核糖核酸(RNA)、肽核酸(PNA)、吗啉代和锁核酸(LNA)、甘油核酸(glycol nucleic acid,GNA)、苏糖核酸(TNA)、单链DNA(ssDNA)、2'-氟脱氧核糖核酸、适体等。
“活化的核酸部分(activated nucleic acid moiety)”是指下述核酸部分,其具有对一种或多种天然官能团的反应性提高的基团。例如,当天然官能团为赖氨酸上的胺时,活化的基团可以是活化的酯,如N-羟基琥珀酰亚胺酯(NHS-酯)。可以将活化的酯直接连接到活化的核酸部分中的核酸组份,或者通过接头连接。当天然官能团为半胱氨酸时,活化的基团可以是马来酰亚胺。其他活化的酯和活化的基团对于活化的核酸部分是有用的。可以使用重氮盐、亚胺和烯丙基钯物质来修饰酪氨酸。可以使用金属卡宾体和亚胺来修饰色氨酸。可以通过氨基转移作用修饰N-末端氨基酸,并且可以使用高碘酸钠氧化N-末端丝氨酸和苏氨酸以获得醛。
“修饰的天然官能团”是指修饰成使得核酸部分结合到细胞表面的天然官能团。可以通过除代谢工程以外的多种方法修饰天然官能团。例如,当天然官能团为糖时,可以使用适合的修饰剂或氧化剂(如高碘酸钠)来氧化糖。当天然官能团为糖并且修饰剂为如高碘酸钠的氧化剂时,所述修饰的天然官能团可以是醛或酮。
“基底表面”是指可以衍生以包含核酸部分的任何材料。用于基底表面的材料的实例包括(但不限于)玻璃(包括可控孔度玻璃)、聚合物(例如聚苯乙烯、聚氨脂、聚苯乙烯-二乙烯基苯共聚物)、硅酮橡胶、石英、乳胶、可衍生化的过渡金属、磁性材料、二氧化硅、氮化硅、砷化镓及其衍生物。除了表面上的反应位点外,所述材料一般能够耐受其可能会经受的多种化学反应条件。
III.细胞-DNA缀合物
本发明提供了细胞-DNA缀合物和制备所述缀合物的方法。
A.无细胞壁细胞的缀合物
本发明提供了核酸部分和无细胞壁细胞的缀合物。通过细胞表面上的天然官能团将核酸部分共价连接到细胞表面上从而形成了缀合物。在本发明所述的缀合物中使用的细胞可以是任何细胞,并且不需要代谢工程来引入用于缀合核酸部分的官能团。
在一些实施方案中,本发明提供了细胞和核酸部分的缀合物,其中所述细胞具有包含天然官能团的表面,所述细胞没有细胞壁,并且所述核酸部分与所述天然官能团共价连接。
在本发明中有用的细胞包括任何细胞类型。在一些实施方案中,所述细胞为无细胞壁细胞。可以用于核酸部分连接的细胞(和用于相同用途的细胞膜)可以是任何真核细胞,其包括所有动物细胞、植物细胞、藻类细胞、细菌细胞和真菌细胞。以下是可以在本发明所述组合物、装置和方法中使用的细胞的非穷举性列表。该列表旨在包括多种细胞和细胞类型,但是不意欲作为可以在本发明中使用的细胞的限制。更确切地,这些细胞和细胞类型是示例性和说明性的。可以在本发明中使用活细胞、已杀死的细胞和细胞系。活细胞可能提供最具有分析性和最有用的信息,并且由于可以在不通过细胞表面修饰进行活化的情况下在本发明中使用活细胞,因此可以使用本发明所提供的和固有的工具在人工系统和装置中研究它们的天然或近天然特性。
以下是细胞的非限制性列表。来自血液和免疫系统的人细胞类型包括:淋巴:B细胞、T细胞(细胞毒T细胞、自然杀伤T细胞、调节性T细胞、T辅助细胞)、自然杀伤细胞,脊髓:粒细胞(嗜碱性粒细胞、嗜酸性粒细胞、嗜中性粒细胞/多叶核中性粒细胞)、单核细胞/巨噬细胞、红细胞(网织红细胞)、肥大细胞、血小板/巨核细胞、树突细胞。内分泌系统包括甲状腺(甲状腺上皮细胞、滤泡旁细胞)、甲状旁腺(甲状旁腺主细胞、嗜酸性细胞)、肾上腺(嗜铬细胞)、松果体细胞(松果体细胞)。神经系统细胞为胶质细胞(星形细胞、小神经胶质细胞)、大细胞性神经分泌细胞、星状细胞、伯特歇尔细胞和垂体(促性腺细胞、促肾上腺皮质细胞、促甲状腺细胞、亲躯体细胞、催乳激素细胞)。呼吸系统细胞包括肺细胞(I型肺细胞、II型肺细胞)、克拉细胞、杯状细胞、尘细胞。循环系统细胞包括心肌细胞、周细胞。消化系统细胞包括胃(胃主细胞、壁细胞)、杯状细胞、帕内特细胞、G细胞、D细胞、ECL细胞、I细胞、K细胞、S细胞。肠内分泌细胞、肠嗜铬细胞、APUD细胞,肝(肝细胞、肝巨噬细胞)、软骨/骨/肌肉。被皮系统,骨:成骨细胞、骨细胞、破骨细胞、齿(成牙骨质细胞、成釉细胞),软骨:成软骨细胞、软骨细胞,皮肤/毛发:丝胞、角化细胞、黑素细胞(痣细胞),肌肉:肌细胞,其他:脂肪细胞、成纤维细胞、腱细胞。泌尿系统包括足状突细胞、近肾小球细胞、球内系膜细胞/球外系膜细胞、肾脏近端肾小管刷状缘细胞、致密斑细胞。生殖系统包括雄性(精子、塞尔托利细胞、睾丸间质细胞)、雌性(卵细胞)。角化上皮细胞包括表皮角化细胞(分化表皮细胞)、表皮基细胞(干细胞)、手指甲和脚趾甲角化细胞、甲床基细胞(干细胞)、髓质毛干细胞、皮质毛干细胞、角质毛干细胞、角质毛根鞘细胞、赫胥黎氏层毛根鞘细胞、汉勒氏层毛根鞘细胞、外毛根鞘细胞、毛基质细胞(干细胞),角膜、舌、口腔、食管、肛管、远段尿道和阴道的复层扁平上皮的湿润复层障壁上皮细胞、表面上皮细胞,角膜、舌、口腔、食管、肛管、远段尿道和阴道的上皮的基细胞(干细胞)、泌尿上皮细胞(粘膜膀胱和尿道)、外分泌上皮细胞、唾液腺粘液细胞(富含多糖的分泌)、唾液腺浆液细胞(富含糖蛋白酶的分泌)、舌中的冯埃布纳腺细胞(清洗味蕾)、乳腺细胞(乳汁分泌)、泪腺细胞(泪液分泌)、耳中的耵聍腺细胞(蜡分泌)、外泌汗腺暗细胞(糖蛋白分泌)、外泌汗腺明细胞(小分子分泌)。顶泌汗腺细胞(体味分泌,性激素敏感)、眼睑中的睫毛腺细胞(特殊汗腺)、皮脂腺细胞(富含脂肪的皮脂分泌)、鼻的鲍曼氏腺细胞(清洗嗅上皮)、十二指肠的十二指肠腺细胞(酶和碱性粘液)、精囊细胞(分泌精液成分,包括精子游动所需的果糖)、前列腺细胞(分泌精液成分)、尿道球腺细胞(粘液分泌)、巴多林氏腺细胞(阴道润滑物质分泌)、利特雷氏腺细胞(粘液分泌)、子宫内膜细胞(碳水化合物分泌)、呼吸道和消化道的离体杯状细胞(粘液分泌)、胃膜粘液细胞(粘液分泌)、胃腺产酶细胞(胃蛋白酶原分泌)、胃腺泌酸细胞(盐酸分泌)、胰腺腺细胞(碳酸氢盐和消化酶分泌)、小肠的帕内特细胞(溶菌酶分泌)、肺的II型肺细胞(表面活性剂分泌)、肺克拉细胞、激素分泌细胞、垂体前叶细胞、亲躯体细胞、催乳激素细胞、促甲状腺细胞、促性腺细胞、促肾上腺皮质细胞、垂体中叶细胞(分泌促黑素细胞激素)、大细胞神经分泌细胞(分泌缩宫素、分泌加压素)、肠道和呼吸道细胞(分泌血清素、分泌内啡肽、分泌促生长素抑制素、分泌胃泌激素、分泌肠促胰液素、分泌缩胆囊肽、分泌胰岛素、分泌高血糖素、分泌铃蟾肽)、甲状腺细胞、甲状腺上皮细胞、滤泡旁细胞、甲状旁腺细胞、甲状旁腺主细胞、嗜酸性细胞(Oxyphilcell)、肾上腺细胞、嗜铬细胞(分泌类固醇激素(盐皮质素类和糖皮质激素))、分泌睾酮的睾丸莱氏细胞、分泌雌激素的卵泡内膜细胞、分泌黄体酮的破裂卵泡的黄体细胞、颗粒黄体细胞、膜黄体细胞、近肾小球细胞(肾素分泌)、肾的致密斑细胞、代谢和储藏细胞、屏障功能细胞(肺、肠、外分泌腺和泌尿生殖道)、肾、I型肺细胞(作为肺气腔的膜)、胰管细胞(泡心细胞)、(汗腺、唾液腺、乳腺等的)无横纹管道细胞、(精囊、前列腺等的)管道细胞、作为封闭的内体腔的膜的上皮细胞、具有推进功能的纤毛细胞、细胞外基质分泌细胞、收缩细胞。骨骼肌细胞、干细胞、心肌细胞、血液和免疫系统细胞、红血球(红细胞)、巨核细胞(血小板前体)、单核细胞、结缔组织巨噬细胞(多种类型)、表皮朗格汉斯细胞、破骨细胞(在骨中)、树突细胞(在淋巴组织中)、小神经胶质细胞(在中枢神经系统中)、嗜中性粒细胞、嗜酸性粒细胞、嗜碱性粒细胞、肥大细胞、辅助T细胞、抑制性T细胞、细胞毒性T细胞、自然杀伤T细胞、B细胞、自然杀伤细胞、网织红细胞、血液和免疫系统的干细胞和定型祖代(多种类型)、多潜能干细胞、全能性干细胞、诱导性多潜能干细胞、成熟干细胞、神经系统、感觉转导细胞、植物神经元细胞、感觉器官和周围神经元支持细胞、中枢神经系统神经元和胶质细胞、透镜状细胞、色素细胞、黑素细胞、视网膜色素上皮细胞、生殖细胞、卵原细胞/卵母细胞、精细胞、精母细胞、精原细胞(精母细胞的干细胞)、精子、营养细胞、卵泡细胞、塞尔托利细胞(睾丸中)、胸腺上皮细胞、间质细胞或肾间质细胞。
所述细胞可以是健康细胞或患病细胞。例如,所述细胞可以来自癌症状况(如上皮癌或上皮瘤,其包括(但不限于)前列腺癌、乳腺癌、结肠癌、胰腺癌、肺癌、皮肤癌(黑素瘤)、食道癌等)或推定的细胞来源(肝细胞癌、肾细胞癌和小细胞肺癌等)。其他癌细胞包括肌上皮癌、肉瘤、神经胶质瘤、淋巴瘤、白血病、类癌瘤以及任何其他类型的癌。可以使用处于其他状态或状况的组织细胞,其包括(但不限于)自体免疫状况、免疫系统相关状况(例如过敏症、对攻击的可能免疫应答)、代表贡献于标准疗法或对其表现出抗性之状况的细胞、对状况的易感性或易患性(例如,对糖尿病、甲状腺状况、中风、心血管状况或肝质量、功能和退变等的易感性)。
在一些实施方案中,所述细胞是原代细胞。在另一些实施方案中,所述细胞是哺乳动物细胞。在另一些实施方案中,所述细胞是干细胞。
所述细胞表面可包含任何适合的天然官能团,如氨基酸和糖。在一些实施方案中,天然官能团可以是氨基酸,如赖氨酸、半胱氨酸、酪氨酸、苏氨酸、丝氨酸、天冬氨酸、谷氨酸或色氨酸。在另一些实施方案中,天然官能团为赖氨酸。在另一些实施方案中,天然官能团可以是N-末端丝氨酸或苏氨酸。
所述核酸部分可以是具有核酸或核苷酸的任何适合的核酸部分。示例性核酸部分包括(但不限于)寡核苷酸、脱氧核糖核酸(DNA)、核糖核酸(RNA)、肽核酸(PNA)、吗啉代和锁核酸(LNA)、甘油核酸(GNA)、苏糖核酸(TNA)、单链DNA(ssDNA)、适体等。其他核酸部分包括氟化核酸。
在另一个实施方案中,本发明所述的核酸部分为核酸以及核苷酸聚合物。术语“核酸”或“寡核苷酸”可以替换使用并且表示包括DNA或RNA核苷酸的生物聚合物,其包括单链、双链、三链、支链或无支链的核酸或由部分杂交的短寡核苷酸梯状物、具有二级结构的核酸和/或天然或非天然核苷酸形成的核酸。
在一个实施方案中,单链寡核苷酸提供了通过与另一细胞、基底表面或装置上它的互补链杂交来与细胞连接的机会。
细胞表面上用于连接的单链寡核苷酸的长度范围可以在约4个核苷酸至约200个核苷酸之间。一般地,约12个核苷酸至40个核苷酸之间的长度最适于杂交。通常,将约20至约25个核苷酸的链用于杂交的目的。
连接到细胞表面上的核酸部分的数目可以在约100000个/细胞以上。在一些实施方案中,核酸部分的数目可以为1个核酸部分至约10000或约30000或约50000。所需的核酸部分的数目可以根据如应用和/或细胞类型的因素而改变。在图1b中,显示在每个细胞上设置了多达120000个DNA链。
在一个实施方案中,所述核酸部分为适体。适体是可具有立体结构并且与特定靶标分子结合的寡核酸分子。通常,通过从较大的随机序列池中进行选择来产生适体,但是天然适体也存在于核开关(riboswitch)中。适体可用于基础研究和作为大分子药物用于临床目的。适体可以与核酶(ribozyme)混合从而在其靶标分子存在时自切割。这些化合物分子具有另外的研究、工业和临床应用。DNA或RNA适体是短链核酸部分。适体是已通过重复多轮体外选择或等价地通过SELEX(通过指数富集的配体系统进化)产生以结合多种分子靶标的核酸物质,所述分子靶标如小分子、蛋白质、核酸以及甚至细胞、组织和生物体。适体可用于生物技术和治疗应用,这是因为它们提供了与常用生物分子、抗体相当的分子识别特性。除了它们的差异性识别外,适体提供了优于抗体的优势,这是因为它们可以完全在试管中产生,易于通过化学合成生产,具有所需的储存性质并且在治疗应用中几乎不或不引起免疫原性。适体选择包括称为核开关的基于核酸的遗传调节元件,其具有与人工制备的适体类似的分子识别特性。这类适体是遗传调节的新模式。
智能适体和智能配体的概念发现了具有适体-靶标相互作用的预定平衡常数(Kd)、速率(koff、kon)常数和热力学(ΔH、ΔS)参数的适体。动力学毛细管电泳选择了适体。未修饰适体在血流中快速被清除,半衰期为几分钟至数小时,这主要是由于核酸酶的降解以及肾脏将其从身体中清除,这是适体固有的低分子量所致。未修饰适体的应用目前集中在治疗短暂的状况,如血液凝固或治疗有可能进行局部递送的器官,如眼。在例如体内诊断成象的应用中,这种快速清除作用可以是有利的。实例为正在开发用于癌症成像的腱生蛋白结合适体。对于科学家来说,如2'-氟取代的嘧啶、聚乙二醇(PEG)连接等(均在FDA批准的适体Macugen中使用)的几种修饰是可用的,通过这些修饰能够容易地将适体的半衰期提高至天或甚至是周的时间尺度。
除了基于适体的治疗剂开发外,多位研究人员已开发了被称为蛋白质组学的完整细胞蛋白质谱的诊断技术,和用于区分疾病和健康状况的医学诊断。作为所有体外选择的资源,适体数据库对所有发表的实验编制了条目。在线通过aptamer.icmb.utexas.edu/对此进行查看。AptaBiD或适体辅助生物标志物开发(Aptamer-Facilitated BiomarkerDiscovery)是用于发现生物标志物的技术。AptaBiD是基于辅助生物标志物指数检测的适体的多次产生或细胞上差异分子靶标的适体混合物的。它包括三个主要阶段:(i)多轮差异选择靶细胞生物标志物的适体;(ii)基于适体,从靶细胞中分离生物标志物;和(iii)生物标志物的质谱鉴定。AptaBiD技术的重要特征在于它在发现生物标志物的同时产生合成亲和探针(适体)。在AptaBiD中,开发了针对处于其天然状态和构象的细胞表面生物标志物的适体。除了有利于生物标志物鉴定外,这些适体可以直接用于细胞分离、细胞显像和体内细胞示踪。它们还可以用于调节细胞受体的活性和将不同的试剂(例如siRNA和药物)递送到细胞中。
适体的亲和力和选择性可以与抗体相当。在本发明中,可以在化学或酶促合成期间容易地对适体进行位点特异性修饰以掺入特定的报告物、接头或其他部分。另外,适体的二级结构可设计成用以经历分析物依赖性构象变化,其与特异性布置化学剂的能力相适应,允许进行多种可能的信号转导模式,而无论检测方式是基于光学、电化学或是质谱。
在另一个实施方案中,所述核酸部分是寡核酸序列,其可用作鉴定序列、条形码(barcode)序列、探针、用于杂交的捕获序列、识别序列、基因表达控制序列、基因序列、增强子和/或掺入或来源于天然酶、蛋白质或其他序列的序列。
在一个实施方案中,连接到细胞的所述核酸部分的序列是相同的。在另一个实施方案中,连接到细胞的所述核酸部分的序列可以是不同的。这将使得核酸部分的连接能够用于多个用途。例如,用于在特定位置捕获细胞的捕获核酸部分,和完成特定活动或用途的杂交或活化序列。
在一些实施方案中,所述核酸部分可以是寡核苷酸、DNA、RNA、PNA或适体。在另一些实施方案中,所述核酸部分可以是单链DNA(ssDNA)。在另一些实施方案中,所述核酸部分可以是约10至约100个核酸。在又一些实施方案中,所述核酸部分可以是适体。
本发明所述的核酸部分还可包括接头。在另一个实施方案中,使用细胞连接系统的化学接头将寡核苷酸连接到细胞表面。接下来参见图1A,所述接头利于结合到细胞表面上的细胞部分,如氨基酸、碳水化合物或其他细胞表面部分。在一个实施方案中,所述接头是可以在不首先修饰氨基酸(或细胞表面上的碳水化合物或其他部分)的情况下直接结合到细胞上氨基酸的部分。化学接头位于要连接的寡核苷酸的一端。在一个实施方案中,通过化学接头与细胞表面蛋白上的氨基酸形成键改变了它的性质,并且氨基酸和核酸寡核苷酸之间通过化学接头形成共价键。在一些实施方案中,所形成的共价键为酰胺键或酯键。因此,在连接到氨基酸上的过程中,化学接头通常将改变其性质,形成酰胺、酯或其他的键,以及细胞表面部分也从而成为共价键的一部分。
在一个具体的实施方案中,N-羟基琥珀酸亚胺(NHS)酯是这样一种可能的化学接头,它在存在碳二亚胺的情况下通过羧酸与NHS的反应形成。含有NHS或硫-NHS酯的试剂与亲核试剂反应以释放NHS或硫-NHS离去基团从而形成了酰基化产物。这类酯与巯基或羟基反应形成了酯键或巯基酯键。这两种键均有可能在水环境中水解或与相邻的胺交换以形成酰胺键和NHS离去基团。
在另一个实施方案中,所述化学接头为杂双官能交联剂。在一个实施方案中,所述杂双官能交联剂为NHS-PEO
在另一个实施方案中,具有聚乙二醇(PEG)(还被称为聚氧化乙烯(PEO))间隔物(spacer)的交联剂是具有纯烃间隔臂之试剂的合适替代方案。PEG间隔物改善了试剂和缀合物的水溶性,降低了缀合物聚集的可能性并提高了交联的灵活性,从而降低了对间隔物本身的免疫应答。与含有包含不同PEG链长的异质混合物的典型PEG试剂相比,这些PEO试剂是具有限定分子量和间隔臂长度的均质化合物,从而为交联应用的优化和表征提供了更大的精确性。例如,通过溶解5mg NHS-PEO
在另一个实施方案中,在存在唾液酸和EDC或l-乙基-3-(3-二甲基氨丙基)碳二亚胺盐酸盐(用于结合含有羧酸盐和胺的生物物质的碳二亚胺)的情况下,NHS-马来酰亚胺可以与巯基-寡核苷酸结合并且与氨基酸反应从而在细胞表面上形成了酯键。
可能与寡核苷酸上的接头形成酰胺、酯或其他键的氨基酸包括赖氨酸、半胱氨酸、天冬酰胺、谷氨酸、酪氨酸、色氨酸和丝氨酸。一般地,赖氨酸、半胱氨酸、天冬酰胺、谷氨酸和酪氨酸与NHS-寡核苷酸形成酰胺键,而丝氨酸将与NHS-寡核苷酸形成酯键。其他接头可以形成不同的键。例如,包括马来酰亚胺、二硫键试剂和酰基化方法可用于与细胞表面蛋白上的半胱氨酸形成直接的共价键。可以在天冬酰胺和谷氨酸处使用酰胺偶联以形成酰胺键。可以对细胞表面上的酪氨酸使用重氮偶联、酰基化和烷基化作用以形成酰胺键。有可能的是可以使用任何氨基酸(20种氨基酸或任何非天然氨基酸)来形成将寡核苷酸与细胞表面连接的直接共价键。所述20种氨基酸是异亮氨酸、亮氨酸、赖氨酸、甲硫氨酸、苯丙氨酸、苏氨酸、色氨酸和缬氨酸(必需氨基酸),以及丙氨酸、天冬酰胺、天冬氨酸、半胱氨酸、谷氨酸、谷氨酰胺、甘氨酸、脯氨酸、丝氨酸和酪氨酸(非必需氨基酸)以及精氨酸和组氨酸。
一般说来,现有技术中可用的任何亲和分子与提供对可检测物质的特异性识别的已知配体相结合将在本发明的核酸基团的连接中有所应用。然后可以连接到这些官能团上的这些生物分子的实例包括具有已知结合配体的接头分子,或者亲和分子,包括(但不限于)多糖、凝集素、选择素(selectin)、核酸(单体和寡聚体)、蛋白质、酶、脂类、抗体和小分子,如糖、肽、适体、药物和配体。
在另一个实施方案中,所述连接是共价的。对本发明有用的双官能交联剂将包含能够偶联到两个不同官能标靶(如肽、蛋白质、大分子、半导体纳米晶体或基底)的两个不同反应基团。所述两个反应基团可以是相同或不同的,其包括(但不限于)例如下列的反应基团,如巯基、羧酸、羰基、胺、羟基、醛、酮、活性氢、酯、巯基或光反应性部分。例如,在一个实施方案中,交联剂可以在官能端具有一个胺反应基团和巯基反应基团。可以在本发明中用作连接试剂的杂双官能交联剂的其他实例包括(但不限于):
-胺反应性+巯基反应性交联剂。
-羰基反应性+巯基反应性交联剂。
-胺反应性+光反应性交联剂
-巯基反应性+光反应性交联剂
-羰基反应性+光反应性交联剂
-羧酸反应性+光反应性交联剂
-精氨酸反应性+光反应性交联剂
以下是交联剂通常所适合的类别的列表。该列表是示例性的并且不应认为是可以用于本发明的交联剂的全部类型。对于每种类别(即这些化学剂所靶向官能团),由于一种反应基团能够与几种官能团反应,因此存在一些子类别。
具有反应基团的大多数交联剂可以广泛地分为以下几类:
1.胺反应性:与含胺(NH2)分子偶联的交联剂。
2.巯基反应性:与含巯基(SH)分子偶联的交联剂。
3.羧酸盐反应性:与含羧酸(COOH)分子偶联的交联剂。
4.羟基反应性:与含羟基(-OH)分子偶联的交联剂。
5.醛和酮反应性:与含醛(-CHO)或酮(R
6.活性氢反应性。
7.光反应性。
更具体地,归入这些类别的化学剂包括(但不限于)含有下列的那些:
1.异硫氰酸酯、异氰酸酯、酰基叠氮、NHS酯、磺酰氯、醛和乙二醛、环氧化物和环氧乙烷、碳酸酯、芳基化剂、亚胺酯、碳二亚胺、酸酐、炔。
2.卤代乙酰基和卤代烷衍生物、马来酰亚胺、氮丙啶、丙烯酰基衍生物、芳基化剂、巯基-二硫化物交换剂。
3.重氮烷和重氮乙酰基化合物,如羰基二咪唑和碳二亚胺。
4.环氧化物和环氧乙烷、羰基二咪唑、高碘酸盐氧化、N,N'-二琥珀酰亚胺基碳酸酯或N-羟基琥珀酰亚胺基氯甲酸酯、酶促氧化、烷基卤素、异氰酸酯。
5.用于形成席夫碱或还原胺化的肼衍生物。
6.用于曼尼希缩合反应和碘化反应的重氮衍生物。
7.芳基叠氮化物和卤化芳基叠氮化物、二苯甲酮、重氮化合物、双吖丙啶衍生物。
对于这些子类别的每一种来说,存在多个化学剂的实例。在现有技术中说明了所有这些化学剂和上述子类别的列表,但是可以在“生物缀合技术(BioconjugateTechniques)”,Greg T Hermanson,Academic Press,San Diego,1996中查到多种这些化学剂,该文献作为参考并入本文。
在其中进行细胞缀合和连接的缓冲溶液的选择取决于对化学接头或交联剂以及对细胞维持生长的条件(即防止细胞裂解)的选择。在一个优选的实施方案中,缓冲溶液的范围在pH6-8之间并且不应含有与化学接头中所使用的与单链核酸反应的官能团相同的官能团。pH7.2是中间pH,但该pH不必是中性的,其取决于与化学反应和细胞状况的相容性。
在一个实施方案中,缓冲溶液是中性pH的磷酸盐缓冲溶液,从而使N-羟基琥珀酰亚胺(NHS)酯(例如NHS-PEO-马来酰亚胺)可用作化学接头。所述反应一般是在允许化学接头与核酸缀合以及随后与细胞或细胞表面连接的条件下进行。在其中使用NHS酯交联剂和磷酸盐缓冲溶液的一些实施方案中,在中性pH(例如pH7.2)和室温下将所述反应进行指定的一段时间(例如,1、3、5、10、15、20、30、45、60分钟或以上)。
在一些实施方案中,缀合物包含在细胞表面上具有赖氨酸的哺乳动物细胞,并且通过酰胺将单链DNA共价连接到赖氨酸上。
可通过本领域已知的任何适合方法制备本发明所述的缀合物。一般说来,所述方法包括将DNA与细胞的天然官能团相连接,其中所述细胞是未修饰的,但是从可能干扰结合步骤的其他生物材料中分离所述细胞。可以使用任何生物缀合技术,如上文在Hermanson中所述的那些技术。
在一些实施方案中,本发明包括通过将细胞与活化的核酸部分相接触来制备细胞和核酸部分的缀合物的方法,其中所述细胞具有包含天然官能团的表面并且其中所述细胞没有细胞壁,从而使所述核酸部分共价连接到所述天然官能团。
如上所述,活化的核酸部分包含接头。在一些实施方案中,活化的核酸部分包括活化的酯。在另一些实施方案中,制备缀合物的方法包括将哺乳动物细胞与活化的核酸部分接触,其中所述天然官能团包含赖氨酸并且所述活化的核酸部分包含NHS酯,从而使所述核酸部分通过形成酰胺键与所述天然官能团共价连接。
B.具有细胞壁的细胞的缀合物
本发明还提供了核酸部分与具有细胞壁的细胞的缀合物以及制备方法。在一些实施方案中,本发明提供了具有细胞壁的细胞和核酸部分的缀合物,其中所述核酸部分共价连接到细胞上。在另一些实施方案中,所述核酸部分共价连接到所述细胞表面上。
天然官能团可以是任何适合的天然官能团,如氨基酸或糖。可用于连接至核酸部分的糖包括(但不限于)葡萄糖、核糖、果糖、唾液酸、甘露糖、半乳糖、蔗糖、乳糖等。其他糖包括(但不限于)单糖、二糖和多糖。
在另一个实施方案中,可以用植物细胞、细菌细胞、真菌、酵母、藻类和古细菌完成寡核苷酸与细胞表面的连接。这种情况下,连接首先需要修饰细胞表面上的碳水化合物分子,然后将寡核苷酸-接头与经修饰的部分连接。动物细胞与植物细胞之间的主要差别在于植物细胞具有细胞壁,因此在寡核苷酸连接前需要修饰。在一个实施方案中,可以通过高碘酸盐氧化然后形成腙来实现寡核苷酸的连接。在一个实施方案中,可以修饰植物细胞上的碳水化合物而不是如在使用哺乳动物或动物细胞的另一些实施方案中一样来修饰蛋白质。在本发明的实施例中,将碳水化合物糖氧化以产生官能化的醛(或酮)。然后,将醛或酮与合成的酰肼-DNA反应以形成被称为腙的共价键。
更一般来说,所述组合物包含缀合至化学接头的寡核苷酸,所述接头共价连接到细胞的外表面。所述键或连接一般是腙、肟或胺。在寡核苷酸与细胞表面碳水化合物之间形成键的方法包含修饰非动物细胞表面碳水化合物以形成醛或酮。这种形成过程可以包括高碘酸盐氧化。然后,将醛或酮与具有官能团的寡核苷酸相接触,所述官能团发生反应形成共价键。所述共价键可以是寡核苷酸上的接头和细胞表面上的碳水化合物之间的腙、肟或胺键。一般地,通过这些键的连接位于细胞表面上的碳水化合物。可以用非动物细胞或动物细胞进行该反应,优选地用非动物细胞进行,如植物、酵母、细菌和藻类以及其他单细胞有机体。在一些实施方案中,所述细胞为植物细胞。
在另一些实施方案中,所述天然官能团包括经修饰的天然官能团。如上所述,所述天然官能团可以是具有1,2-二醇基的糖,所述1,2-二醇基氧化形成醛或酮。在另一些实施方案中,经修饰的天然官能团包括氧化的糖。在又一些实施方案中,经修饰的天然官能团包括唾液酸、甘露糖、葡萄糖、半乳糖、N-乙酰葡萄糖胺或N-乙酰甘露糖胺。
本发明还提供了制备核酸部分与具有细胞壁的细胞的缀合物的方法。如上所述,该方法可包括两步的过程:首先修饰具有细胞壁的细胞的天然官能团,然后将核酸部分缀合到经修饰的天然官能团。
在一些实施方案中,本发明提供了制备细胞与核酸部分的缀合物的方法,其包括将所述细胞与活化的核酸部分相接触,其中所述细胞具有细胞壁,从而使所述核酸部分与所述细胞相连接。所述活化的核酸部分可包含任何适合的活化基团以使得所述核酸部分与所述细胞相缀合,如上所述。在一些实施方案中,所述活化的核酸部分包括氨基氧、酰肼、肼、氨基脲、氨基硫脲或胺。作为另外一种选择,活化的核酸部分可包含半胱氨酸以形成噻唑烷或包含丝氨酸以形成噁唑烷。在Hermanson(参见上文)中描述了将核酸部分与细胞缀合的其他方法。
用具有细胞壁的细胞制备缀合物的方法还包括修饰天然官能团的步骤。在一些实施方案中,制备核酸部分与具有细胞壁的细胞的缀合物的方法包括将天然官能团与修饰剂相接触以制备经修饰的天然官能团,从而使核酸部分能够共价连接到经修饰的天然官能团上。
所述修饰剂可以是任何适合于制备经修饰天然官能团的试剂。例如,所述修饰剂包括(但不限于)氧化剂。在一些实施方案中,所述修饰剂包括氧化剂。适合的氧化剂包括(但不限于)高碘酸钠。在另一些实施方案中,所述氧化剂包括高碘酸钠。在另一些实施方案中,经修饰的天然官能团包括氧化的糖。在又一些实施方案中,经修饰的天然官能团包括氧化的唾液酸。在又一些实施方案中,经修饰的天然官能团包括醛基。
IV.装置
本发明还提供了包含本发明所述缀合物的基底表面和装置。在一些实施方案中,本发明提供了包含具有细胞表面的细胞的装置,所述细胞表面包含共价连接到第一核酸部分的天然官能团。所述装置还包含基底表面,其包含与第一核酸部分互补的第二核酸部分,从而通过形成第一和第二核酸部分的核酸双链体将细胞结合到基底表面上。
在一些实施方案中,所述细胞为动物细胞。
所述基底表面可以是任何适合的材料。适合材料的实例包括(但不限于)玻璃(包括可控孔度玻璃)、聚合物(例如聚苯乙烯、聚氨脂、聚苯乙烯-二乙烯基苯共聚物)、硅酮橡胶、石英、乳胶、可衍生化的过渡金属、磁性材料、二氧化硅、氮化硅、砷化镓及其衍生物。基底表面还可以具有任何适合的表面几何形状,其包括(但不限于)平面,曲线和球形。
在一些实施方案中,所述基底表面是平面。在另一些实施方案中,所述基底表面是球面。
在一些实施方案中,所述装置还包括流体通道。在另一些实施方案中,通道为微通道。在又一些实施方案中,通道为纳米通道。
在一些实施方案中,所述装置包含传感器。在另一些实施方案中,所述传感器包括纳米传感器。在另一些实施方案中,所述传感器包含电极。在又一些实施方案中,所述传感器包括压电传感器。在另一个实施方案中,所述装置适于原子力显微镜检查。
在一些实施方案中,所述装置适于细胞的生物化学或电化学分析。在另一些实施方案中,生物化学分析包括基因组分析。在另一些实施方案,所述装置还包含选自下列的组件:微制造加热器、温度传感器、聚合酶链反应(PCR)室或毛细管电泳分离通道。在又一些实施方案中,装置能够产生细胞的转录谱。
在一些实施方案中,所述装置还包括生物反应器。
在另一个实施方案中,本发明提供了平台,其用于观察和筛选可以通过由寡核苷酸连接所引起的细胞捕获进行研究的过程,如伤口愈合、组织再生、感染、对测试药物的反应性或响应性、一般性药物筛选和对刺激物响应的基因表达。本发明还提供了方法和综合系统,其允许通过寡核苷酸连接所致细胞捕获对多种人状态和状况(例如患病的和正常的)进行研究和检测。例如,还可以在通过检测细胞表面标志物与捕获寡核苷酸的杂交来检测疾病细胞表面标志物的疾病诊断实践中应用寡核苷酸与细胞的连接。
在其表面上具有连接的寡核苷酸链的细胞可以在三维空间(如流体或凝胶)中彼此杂交或退火。该技术的多种应用是可能的。与在平面上可以展示的相比,三维支架或网可用于使多得多的细胞图案化。因此,在一个实施方案中,可对网、多孔大块材料或支架进行修饰以展示连接到表面上的寡核苷酸从而使通过本发明方法修饰的细胞可展示或捕获在表面上。这些表面可用于研究遗传学、衰老和药物应答或者用于细胞副产物的代谢工程、生产和收集。
例如,细胞可以用于研究组织再生,如心肌组织再生,其中在累积足够数目的细胞后,临界数目的细胞形成搏动单元。使用接种的细胞连接基质,潜在地可“培养”任何组织,以产生相互作用细胞的网络。可以研究和控制这些细胞聚集体和模拟组织的动态和产物。例如,可以使用细胞连接系统来辅助干细胞的分化和储藏。例如,成熟干细胞和诱导性多潜能干细胞可保持不分化或导向为所需细胞类型的分化状态。可以产生人工组织以替换包括人在内的动物中的缺失、受损或患病的组织。可以在适当的环境中体内或体外实施神经和脊柱组织再生。为了指导或研究该系统中的细胞,可以将传感器布置在细胞内或布置在与细胞接触的表面上。
所述细胞连接系统可用于递送目的,其使用具有连接的寡核苷酸的细胞来递送其他细胞表面分子,如适体、基因调节分子、基因表达控制分子、控制基因表达的基因。所述细胞连接系统还可用于将具有连接的寡核苷酸的细胞的内容物递送至另一个细胞。例如,小分子、肽、肽模拟物、具有用于基因表达的DNA的载体、如小抑制RNA或小发夹RNA的干扰RNA分子和其他可递送的分子。细胞表面上的适体可以结合内化受体,该细胞与其内容物一起内化。一般地,在其表面上具有寡核苷酸(即适于结合蛋白质的适体)的细胞提供了递送到或进入另一个活细胞的机制。适体本身可包含适于在细胞内起作用的核酸,如用于在细胞中表达的寡核苷酸、干扰RNA(如siRNA、shRNA或微小RNA)或分子信标,如具有适于结合细胞中特异性序列的标记物的核酸,并且提供对该序列或该序列所表示基因的光学探测。在其他基因组修饰活性中,所述核酸可提高基因表达、控制基因表达、阻断基因表达或改变基因表达。
在另一个实施方案,本发明提供了用于在基底上对细胞进行序列特异性图案化或捕获的方法以及筛选方法。例如,如实施例中所述,可以将细胞或细胞样品与PBS磷酸盐缓冲液(pH7.2)以及接头一起孵育几分钟至几小时以将单链寡核苷酸连接到细胞表面上,由此产生用寡核苷酸在其表面上进行了修饰的细胞。可以制备具有连接到基底表面上的序列(例如,ssDNA捕获序列)的基底表面。在一个实施方案中,连接到细胞上的单链寡核苷酸与连接到基底上的ssDNA捕获序列互补从而使得当允许细胞接触基底时,两个序列杂交,因此将细胞固定在基底上。
因此,在另一个实施方案中,本发明还提供了具有以下特征的装置,使用本发明的细胞修饰方法将细胞固定在表面上。在一个实施方案中,本发明的装置可以是在微阵列或者微阵列样的图案中图案化的传感器。在另一个实施方案中,可以在分析细胞或其他目的的装置中使用具有连接的寡核苷酸的细胞或细胞膜。在一些实施方案中,所述装置将在其表面上具有互补寡核苷酸以用于与使用本发明方法连接到细胞上的寡核苷酸杂交。所述装置可以是任何有用的形状,例如,平面或球形。它可以具有流体通道(即微通道或纳米通道)。所述装置可包括传感器,如纳米传感器、电极或压电电极。所述装置可适于原子力显微镜检查、细胞生物化学分析或基因组分析。所述装置可具有其他组件,如加热器、温度传感器和如在其他可能的方法中用于产生细胞转录谱的PCR室和毛细管电泳分离通道的组件。所述装置还可包含生物反应器。所述装置可包括选自微制造的加热器、温度传感器、聚合酶链反应(PCR)室和毛细管电泳分离通道的组件。
在一个实施方案中,为了检测杂交事件,根据所述装置,对装置上与细胞连接的寡核苷酸或互补寡核苷酸或捕获寡核苷酸进行标记。例如,如果将标记物与扩增混合物一起加入,则cDNA位于模板链而探针位于有义链(除非它们是阴性对照)。标记物通常是荧光的,尽管偶而会使用放射性标记等。标记可以是直接或间接的。间接标记需要有偶联阶段,其可以在杂交之前或之后发生。如果在杂交前发生标记,则可以使用通过染料(如氨基烯丙基-UTP和NHS氨基-反应性染料如花青染料)标记的杂交核苷酸(例如在双通道阵列中)。氨基烯丙基基团是通常位于连接到核碱基的长接头上的胺基,它与反应性染料反应。在一些实施方案中,以与正常核苷酸相比较低的比例酶促地加入经修饰的核苷酸(通常1个aaUTP:4个TTP混合物),从而通常导致每60个碱基中有1个,如用分光光度计所测量的。然后,例如,使用含有Tris-磷酸缓冲液(含胺基)的溶液,用色谱柱纯化aaDNA。
可以使用多种技术制造微阵列,其包括用尖端针在载玻片上印制,使用预制掩模的光刻法,使用动态微镜装置的光刻法,喷墨印刷或微电极阵列上的电化学法。在点制的微阵列中,探针是对应于mRNA的寡核苷酸、cDNA或小片段的PCR产物。在沉积到阵列表面上之前合成探针,然后将该探针“点样”到玻璃上。常用方法使用了机械手控制的细针或针阵列,其浸入到含有DNA探针的孔中,然后将每种探针沉积在阵列表面上的指定位置处。所得的探针“网格”代表所制备探针的核酸谱并且准备接收来源于实验或临床样品的互补cDNA或cRNA“靶标”。
在一个实施方案中,这些寡核苷酸微阵列中的寡核苷酸探针是设计成与已知或预测的可读框的序列部分相匹配的短序列。在该阵列中,可以通过直接在阵列表面上合成该序列而不是沉积完整序列来印制设计成表示单个基因或基因剪接变体家族的短寡核苷酸序列,从而产生基底上排成阵列的寡核苷酸。根据所需的目的,序列可以较长(60聚体探针,如Agilent的设计)或较短(Affymetrix生产的25聚体探针);较长的探针对个体靶标基因的特异性更强,而较短的探针可以以更高的密度在阵列上点样,并且生产成本更低。用于生产寡核苷酸阵列的一种技术包括在二氧化硅基底上的光刻合成(Agilent和Affymetrix),其中使用光和光敏掩蔽剂在整个阵列上每次一个核苷酸地“构造”序列。在将阵列浸入单个核苷酸的溶液中之前,将每个适用的探针选择性地“去掩蔽”,然后发生掩蔽反应,将下一组探针去掩蔽以准备不同核苷酸的暴露。多次重复后,完全构造了每个探针序列。最近,NimbleGen Systems的无掩蔽阵列合成(Maskless Array Synthesis)将灵活性与大数量的探针相结合。
双色微阵列或双通道微阵列通常与从待比较的两个样品(例如,患病组织与健康组织或者患病细胞与健康细胞)中制备的并且用两种不同的荧光团标记的cDNA杂交。一般用于cDNA标记的荧光染料包括荧光发射波长为570nm(相当于光谱中的绿光部分)的Cy3和荧光发射波长为670nm(相当于光谱中的红光部分)的Cy5。将两种Cy标记的cDNA样品混合并与单个微阵列杂交,然后将所述微阵列在用限定波长的激光束激发后在微阵列扫描器中扫描从而使两个荧光团的荧光成像。然后,可以在基于比值的分析中使用每个荧光团的相对强度以鉴定上调和下调的基因。
可以对细胞实施本发明的细胞修饰并且可以在微流体应用、方法和装置中使用。本发明的细胞表面修饰方法使得能够在表面上捕获和固定单个细胞,借此允许以多种方式对细胞作用。例如,在一个具体的实施方案中,可以制备集成微流体装置,如在实施例4中所述的和在Toriello等人,Integrated microfluidic bioprocessor for single-cellgene expression analysis,Proc.Natl.Acad.Sci.U.S.A.,105,20173-20178,2008Dec23;105(51):20173-8,Epub 2008Dec 15中所述的,上述参考文献作为参考并入本文。开发了集成微装置以用于分析单细胞中的基因表达。通过在该集成微装置表面上固定和捕获单细胞,使得能够进行单细胞的逆转录PCR扩增。
因此,在一个实施方案中,本发明方法实现了以使用本发明的直接细胞修饰的固定化细胞为特征的微流体结构。这些微流体结构可以包括微气动系统,即,用于操纵芯片外流体的微系统(液体泵、气阀等)和用于在芯片上操纵纳升或皮升体积以在多种分子生物学过程中使用的微流体结构,所述分子生物学过程如酶学分析(例如葡萄糖和乳酸测定)、DNA分析(例如聚合酶链式反应和高通量测序)和蛋白质组学。
这些微流体装置旨在集成测定操作(如检测)以及在一块芯片上进行样品预处理和样品制备。生物芯片新兴的应用领域有临床病理学,特别是疾病的即时现场诊断。另外,能够连续采样并且实时测试空气/水样的生物化学毒素和其他危险病原体的基于微流体的装置可以用作早期报警的生物传感器(例如“生物烟气报警器”)。
在另一个实施方案中,用于具有通过本发明方法连接的寡核苷酸之细胞的微流体包括连续流动技术,其基于对通过微制造通道流体的连续液体的操纵。通过外部压力源、外部机械泵、集成机械微型泵或通过电动机构实施了液流动作。由于易于实施并且对蛋白质污染问题不太敏感,因此连续流动微流体操作是主流方法。连续流动装置适于多种明确限定的且简单的生物化学应用以及如化学分离的某些工作,但是它们不太适合于需要高度灵活性或复杂液体操纵的工作。基于提供低至纳升范围的分辨率的MEMS技术,可以通过高灵敏度的微流体流动传感器实现连续流动系统中的过程监测能力。
另一些实施方案包括(但不限于)上述封闭通道连续流动系统的替代性数字(基于液滴)微流体系统,其包括新型开放结构,其中在基底上操纵分散、可独立控制的液滴。
除了微阵列外,另一种实施方案包括设计用于双向电泳、转录组分析、PCR扩增等的生物芯片。其他应用包括用于蛋白质和DNA的多种电泳和液相色谱应用、细胞分离(特别是血细胞分离)、蛋白质分析、细胞操纵和分析,包括细胞存活分析和微生物捕获。
在另一个实施方案中,本发明的直接细胞修饰方法使装置能够进行用于单细胞成像、操纵和图案化。例如,可以使用原子力显微镜(AFM)或扫描力显微镜(SFM)分析细胞或使细胞图案化。因此,在一个实施方案中,在Hsiao SC等人,DNA-coated AFM cantileversfor the investigation of cell adhesion and the patterning of live cells,AngewChem Int Ed Engl.2008;47(44):8473-7,16Sep 2008Epub中(其作为参考并入本文)和在实施例8中说明了如AFM悬臂的装置。本发明中所描述的技术为AFM装置所带来的优势是使用AFM悬臂精确放置细胞用于分析和操纵的能力。AFM和SFM是极高分辨率类型的扫描探针显微镜,其显示出几分之一纳米的分辨率,比光衍射限度高出1000倍以上。
使用本发明的寡核苷酸连接系统,对完整活细胞进行AFM的能力提供了特异性分析多种细胞类型的机会,并且同时,由于通过它的寡核苷酸锚定点稳固地保持和控制,因此的确以更高的保真度分析了单个细胞。例如,AFM的另一个主要应用(除了成像之外)是作用力光谱,即作用力-距离曲线的测量。对于这种方法,由于监测悬臂的静变位(staticdeflection)相对于压电移位的函数,因此AFM尖向表面延伸并从表面收回。这些测量已用于测量纳米尺度的接触、原子键、范德华力和卡西米尔作用力、液体中的溶解力和单分子拉力和断裂力。现在,通常可以用优于0.1纳米的垂直距离分辨率测量几皮牛顿左右的作用力。
微米和纳米技术应用于化学分析、环境监测、医学诊断和用于制药学的细胞组学和微型反应器。通过使用纳米技术,预期对芯片上实验室(lab on a chip,LOC)系统的研究也朝着液体输送结构缩小的方向扩展。能够使亚微米和纳米尺寸通道、DNA迷路(labyrinth)、单细胞检测和分析以及纳米传感器可能会变得可行,其允许生物物质和大分子以新的方式进行相互作用。本发明有助于使这些系统有可能用于整个细胞。LOC系统可实现实时PCR,有利于生物化学测定、免疫测定、基于抗原-抗体反应检测细菌、病毒和癌症、检测癌细胞和细菌的双向电泳、血样制备,可以破裂细胞以提取DNA、细胞分析、通道内筛选。
芯片上实验室技术可能不久将会成为改善全球健康工作的重要部分,特别是通过现场测试装置的开发。在医疗资源较少的国家中,那些在发达国家中可治疗的传染性疾病通常是致命的。在一些情况下,较差的医疗诊所具有治疗某些疾病的药物,但是缺乏诊断工具来鉴别应该接受这些药物的患者。多位研究人员相信LOC技术可能是强大的新型诊断仪器的关键。这些研究人员的目标是产生微流体芯片,其将允许设备条件较差的诊所中的医疗卫生人员在没有实验室支持的情况下能够实施诊断测试(如免疫测定和核酸测定)。已经为现场测试(point-of-care testing,POCT)的临床诊断设计、制造和表征了用于逆转录(RT)-聚合酶链反应(PCR)的新型聚合物芯片上实验室(LOC)。另外,还已开发了由用于RT-PCR过程的基于非接触红外线(IR)的温度控制系统和用于芯片上检测的光学检测系统组成的便携式分析仪,并已用于监测RT-PCR LOC。
在另一个实施方案中,已构建了完全整合的基因组分析微系统,其包括微制造的加热器、温度传感器和直接与毛细管电泳分离通道相连的PCR室。在实施例6中描述了用于单细胞基因表达分析的集成微流体生物处理器,以及在Toriello等人,“Integratedmicrofluidic bioprocessor for single-cell gene expression analysis”,PNAS,December 23,2008,vol.105no.51 20173-20178,9/16/08(在线刊出)中也有描述,该参考文献作为参考并入本文。该装置是通向法医和现场分子医学诊断中使用的微制造的基因组微处理器的重要步骤。无论是在装置上展示细胞还是将细胞彼此连接,可以通过用多种技术测量mRNA水平来确定基因表达分析,所述技术包括微阵列、表达cDNA序列标签(EST)测序、基因表达顺序分析(SAGE)标签测序、大规模平行信号测序(MPSS)或多重原位杂交的多种应用。所有这些技术都极其易受噪音影响的和/或在生物测量中发生偏差,计算生物学的主要研究领域涉及开发用于将高通量基因表达研究中的信号与噪音分离的统计学工具。这些研究通常用于测定与病症有关的基因:可以将癌性上皮细胞的微阵列数据与非癌性细胞的数据进行比较以确定特定癌细胞群体中上调或下调的转录本。
同样,调控分析可以发生在这些寡核苷酸连接的细胞中:调控是从细胞外信号(如激素)开始并导致一种或多种蛋白质活性提高或降低的事件的复杂组合。生物信息学技术已用于探索该过程中的多个步骤。例如,启动子分析包括对基因编码区周围的DNA中序列基序的鉴定和研究。这些基序影响该区域向mRNA转录的程度。表达数据可用于推断基因调控:可以比较来自多种状态的生物的微阵列数据以形成关于与每种状态有关的基因的假设。在单细胞生物中,可以比较细胞周期的阶段以及多种应激条件(热休克、饥饿等)。然后,可以对这些表达数据应用聚类算法以确定共表达的基因。例如,可以对共表达基因的上游区(启动子)搜索过度表现的调控元件。
在另一个实施方案,将连接有寡核苷酸的本发明经修饰细胞用于或集成传感器装置中,所述传感器装置测量物理量并将其转换为观察者或仪器可以读取的信号。例如,还可以使用这些组合物和系统完成蛋白质表达的分析。蛋白质微阵列和高通量(HT)质谱(MS)可以提供细胞中所存在的蛋白质的快照。生物信息学深入涉及对蛋白质微阵列和HT MS数据的解释;前一种方法面临与靶向mRNA的微阵列相似的问题,而后者涉及将大量质谱数据与蛋白质序列数据库中预测的质量数进行匹配的问题以及复杂的样品统计分析,其中检测了来自每种蛋白的多个,但并不完整的肽。
可以使用寡核苷酸捕获的细胞通过这些系统完成癌症或任何其他疾病或状况中突变的分析。在癌症中,受影响细胞的基因组以复杂或甚至是不可预测的方式进行重排。大规模测序工作已用于鉴别癌症的多个基因中先前未知的点突变。生物信息学家不断产生了专门的自动化系统以管理所产生的大量序列数据,并且它们建立了新的算法和软件来将测序结果与人基因组序列不断增长的集合以及种系多态性进行比较。使用了新的物理检测技术,如鉴定染色体增加或丢失(称为比较基因组杂交)的寡核苷酸微阵列和检测已知点突变的单核苷酸多态性阵列。这些检测方法同时测量了整个基因组中几十万个位点,并且当用于高通量地测量数以千计的样品时,每个实验产生了万亿字节的数据。另外,大量和新型的数据为生物信息学家创造了新的机会。通常发现数据包含相当大的变异性或噪音,并且因此正在开发隐马尔可夫(Hidden Markov)模型和变点分析方法以推断真实的拷贝数变化。在生物信息学的结构学分支中,使用同源性来确定在结构形成和与其他蛋白质相互作用中重要的蛋白质部分。在称为同源建模的技术中,一旦已知同源蛋白质的结构,则该信息用于预测蛋白质的结构。这在目前仍是可靠预测蛋白质结构的唯一方法。
使用本发明系统可进行比较基因组学分析。比较基因组分析的核心是在不同生物的基因(直系同源分析)或其他基因组特征之间建立对应性。正是这些基因组间图谱使得有可能对使两个基因组趋异的进化过程进行追踪。在多个组织水平下作用的多个进化事件形成了基因组的进化。在最低的水平下,点突变影响个别核苷酸。在较高的水平下,较大的染色体部分经历了重复、侧向转移、倒位、转座、缺失和插入。在最根本上地,整个基因组参与了通常导致快速物种形成的杂交、多倍体化和内共生过程。基因组进化的复杂性为数学模型和算法的开发者(他们求助于一系列算法、统计学和数学技术,从基于节约原则模型(parsimony model)的针对问题的确切、启发式、固定参数和近似算法到基于概率模型的针对问题的贝叶斯(Bayesian)分析的马尔可夫链蒙特卡洛(Markov Chain Monte Carlo)算法)提供了多个令人兴奋的问题。
所述细胞和系统可用于对患者进行预后和诊断。可使用通过直接细胞修饰而实现的装置对各个基因组进行基因分型和分析。例如,基因分型阶段可以使用多种不同的实验方法,其包括单核苷酸多态性(SNP)芯片(通常基因组的0.02%)或者部分或完全基因组测序。一旦基因型已知,则存在可以比较各个基因组并且发现基因和位点的疾病相关性的多个生物信息学分析工具。
在另一个实施方案中,在生物反应器装置或系统中使用了本发明修饰的细胞以利于细胞中或几个分子间的生物活性,并且一般导致产生了最终产物或其他一些所需的结果,如生物活性(例如,代谢、能量产生、电信号、分解代谢、凋亡、生长、分化、增殖等)。例如,可以在这些系统中研究细胞代谢。生物反应器可以是指支持生物活性环境的任何装置或系统。
生物材料、干细胞、生长和分化因子以及仿生环境方面的科学进步为在实验室中从设计的细胞外基质(“支架”)、细胞和生物学活性分子的组合制造组织创造了独特的机会。在另一个实施方案中,本发明的细胞修饰方法使得细胞能够连接或置于其他表面上,其旨在使细胞或组织在细胞培养物的背景下生长。例如,具有通过共价键连接到细胞表面上的单链寡核苷酸的细胞可以与相邻细胞或表面上的互补链杂交,因此提供了将细胞连接在一起和/或连接到表面上的方法以产生和研究细胞、组织和器官。组织工程的多学科领域中的巨大发展产生了一组新型组织替换部件和实施策略,其可用于本发明。在一个实施方案中,可以将用连接的寡核苷酸修饰的细胞植入或“接种”到能够支持三维组织形成的人工结构中。可以修饰大块材料、支架、框架,其通常对于重演体内环境来说在离体和体内非常关键并且允许细胞影响它们自身微环境,从而允许使用本发明方法连接细胞。
在一个实施方案中,使用本发明方法修饰光合单细胞生物并且连接寡核苷酸。计划使用的生物包括(但不限于),光合生物:莱茵衣藻(C.reinhardtii)(藻类)、集胞藻(Synechocystis)PCC6803(蓝细菌)、深红红螺菌(R.rubrum)(革兰氏阴性厌氧菌);异养生物:巴斯德梭菌(C.pasteurianum)(固氮菌)、固氮菌属(Azotobacter)(提议具有任何生物的最高呼吸率)。(参见Melis等人,在本文的其他部分中所引用的)。
在一个实施方案中,修饰藻并连接寡核苷酸。由于可以快速生长并且可以具有高百分比的脂肪或油脂,因此藻类可以是特别适合的。它们一天可以使质量倍增数次,并且每英亩产生比替代物(如油菜籽、棕榈、大豆或麻风树)多至少15倍的油脂。由于不需要清水,因此藻养殖也比多种其他作物更经济,并且对淡水资源产生小得多的压力。它还可以在不除去粮食作物的情况下生长。寡核苷酸连接系统使得能够控制植物、细菌或藻类细胞在表面上的布置,例如,与表面成一定角度以最大程度地暴露于日光。
在另一个实施方案中,可以在燃料电池或其他电化学转换装置中使用具有连接的寡核苷酸的本发明修饰的细胞。它从燃料(阳极侧)和氧化剂(阴极侧)产生电流,燃料和氧化剂在电解质存在下反应。反应物流入到电池中,反应产物从中流出,而电解质保持在电池内。只要维持必需的流量,那么几乎可以连续运行燃料电池。燃料电池不同于电化学电池,在燃料电池中它们消耗来自外部来源的反应物,其必须补充-热力学开放系统。相比之下,电池通过化学方式储存电能,因此代表了热力学封闭系统。燃料和氧化剂的多种组合是可能的。氢燃料电池使用氢气作为燃料而使用氧(通常来自空气)作为氧化剂。其他燃料包括烃类和醇。其他氧化剂包括氯和二氧化氯。
使用绿藻莱茵衣藻(Chlamydomonas reinhardtii)可以产生活的太阳能电池以用于微生物发电,如Rosenbaum,Appl.Microbiol.Biotechnol.(2005)68:753-756中所述的。在Wang等人,Electrochimica Acta 54(2009)1109-1114中描述了双室微生物燃料电池。在He等人,Environ.Sci.Technol.2009,43,1648-1654中描述了自维持光合自养微生物燃料电池(MFC),其中使用光合微生物和异养细菌的混合微生物群落从自维持沉积物光合自养MFC中产生电流。从这些MFC中恒定地产生电流而不输入有机化合物或营养物。Kjeang等人(Journal of Power Sources 158(2006)1-12)说明了微生物燃料电池的策略性酶图案化,从而相对于个体转换率,优化了非分隔燃料电池组件中的混合的燃料和氧化剂通道,所述电池组件具有在装置中图案化的分开的酶。Ringeisen等人,J.of Power Sources 165(2007)591-597中描述了使用在厌氧和好氧环境中保持活性的沙雷菌(Shewanella(DSP10)oneidensis)构建的微型MFC。先前的研究显示来自该细菌的电子已经在氧存在下用于还原金属。在该装置中,细菌用作阳极室中的活性电化学物质。该论文指出用于水下监视系统的传感器需要可以在好氧环境(即接近水面的水柱中)中工作的小型(隐形的,与它所供电的装置相比在尺寸上不占优势)理想电源,它提供了连续的电能(不存在充电或使用寿命的问题)并且不需要高度的太阳辐照(因此能够在水下和在夜间等工作)。由于阳极不必需处于厌氧环境中,因此该小组选择用于研究的细菌为传感器网络的RF通讯提供了机会,并且因此可以在水面上的光照环境中工作。还报道了如果由太阳能驱动,则可以将当前设计的微生物燃料电池制备得更可靠,并且这些系统中可能的电能取决于氮源的性质和光的可用性。在仔细设计的系统中,可以使用太阳能驱动该系统中的氮处理细胞以驱动氮(例如,来自沉积物、废水或农业废物)处理系统的组合(参见Cho等人,J.Applied Microbiology 104(2008)640),其结果是获得了更多的电流并提高了系统的寿命。
因此,可以制备使用本发明的寡核苷酸连接系统的装置,如(例如)使用本发明的寡核苷酸修饰的非动物细胞以及与本发明相一致的其他原理的染料电池装置。例如,使用经修饰的植物细胞可制备产氢燃料电池。通常,可以使植物细胞图案化从而使产氢者和除氧者以最适布置混合以用于产生能量。装置可以是平面或球形的,或提供对太阳能捕获和处理来说最佳的任何表面并且可以包括传感器。还可以制备装置以研究或为了能量目的而使用植物、细菌或藻类细胞。该装置还可用于储存能量。该装置可包含传感器或者可以是生物反应器。基于藻类的系统可以是使用通过绿藻光合产H
本发明所述工具可以应用的光合、光伏基于非动物细胞的其他系统包括Dickson等人,International J.Hydrogen Energy,34(2009)204-215,其描述了集胞藻(Synechocystis)PCC6803在形成二氧化硅溶胶-凝胶中的用途。此类技术可直接用于驱动小型电子装置。Sui等人,J.of Microelectromechanical systems vol.17,no.65,Dec2008中描述了微制造的聚二甲基硅氧烷(PDMS)微生物燃料电池(MFC),其具有包埋的微柱电极。该MFC的特征在于适于体内植入的灵活性和生物相容性结构,其作为植入bioMEMS装置的可能的电源。Song等人描述了用于bioMEMS装置的使用聚二甲基硅氧烷(PDMS)的微流体聚合物电解质膜(PEM)燃料电池,它可用于驱动便携式电子装置以及其他小型化MEMS装置。Song等人所述的工作使用可逆结合的PDMS微通道使亚微米厚的纳菲(Nafion)膜在玻璃基底上图案化从而在燃料电池电极之间产生了离子选择性膜而不是将纳菲薄片夹在中间。由于PDMS材料的柔韧性,可以通过氧等离子体粘合将纳菲膜密封在PDMS芯片和玻璃基底之间。可以将表面图案无缝地集成到标准微制造工艺流程中并且形成PEM微流体燃料电池,而无需将层夹在一起的繁重工作并且没有由于夹子失效而造成泄漏的固有风险。至少,该装置的构建使得有可能构建大规模平行微流体燃料电池阵列。使用本发明所述的细胞连接方法可利于细胞图案化。
在另一个实施方案中,使用本发明修饰技术修饰的细胞表面是细胞膜样表面,如病毒、噬菌体的衣壳蛋白和其他自组装生物分子表面或结构。实施例9表明通过本发明细胞修饰方法修饰的病毒或噬菌体衣壳将DNA适体与表面结合,从而使得能够产生多价细胞靶向载体。因此,在多种单体或生物分子表面的修饰中,本发明的直接细胞修饰方法可以用于需要核酸或寡核苷酸的直接连接的多种应用。
V.试剂盒
本发明还提供了试剂盒,其具有适于形成本发明的核酸-细胞缀合物的活化的核酸部分,和具有互补核酸部分的基底表面。在一些实施方案中,本发明提供了适于共价连接到细胞表面的天然官能团上的活化的核酸部分,和包含与所述活化的核酸部分互补的核酸部分的基底表面。所述活化的核酸部分可以是粉末或在溶液中。试剂盒还可以包含本领域技术人员所知的缓冲溶液,以及用于形成核酸-细胞缀合物并且然后将所述核酸-细胞缀合物通过基底表面上的核酸部分与基底表面结合的其他溶液。
以上更详细地描述了试剂盒的组件。所述基底表面可包括任何适合的材料,其包括(但不限于)显微镜载玻片、盖玻片、组织培养板或孔板。可以在特定的位置用核酸部分预印制基底表面。试剂盒可包括其他组件,如细胞固定溶液、检测剂溶液、细胞染色剂溶液、缓冲溶液等。其他溶液可包括核酸部分本身,以及用于将核酸部分连接到细胞上的溶液和反应物。
VI.实施例
除非另作说明,否则所有细胞培养试剂均得自Gibco/Invitrogen Corp(Carlsbad,CA)。使用标准技术进行细胞培养。Jurkat细胞在T-25培养瓶(Corning,USA)中在添加了10%(v/v)胎牛血清(FBS,HyClone)和1%青霉素/链霉素(P/S,Sigma)的RPMI培养基1640中生长。MCF-7细胞在添加了1%非必需氨基酸和10%胎牛血清加1%青霉素/链霉素的DMEM中生长。MDA-MB-231细胞在与MCF-7细胞相同但未加入非必需氨基酸的条件下生长。
使用具有DAPI/Hoechst、荧光素/fluo-3和罗丹明的荧光滤镜组的Axiovert 200M倒置显微镜(ZEISS)获取荧光显微照片。使用UVIKON 933双光束UV/vis分光光度计(Kontron Instruments,United Kingdom)在260nm下确定不同寡核苷酸的紫外吸收。
NHS-DNA缀合物的合成。对于细胞粘附研究,设计了三对互补寡核苷酸,使它们在总体组成方面是相同的,仅在序列方面是不同的。还对每对序列进行计算以具有相当的熔链温度(55℃)和最小的二级结构。
序列身份如下:
C1:5′-GTA ACG ATC CAG CTG TCA CT-3′
M1:5′-AGT GAC AGC TGG ATC GTTAC-3′
C2:5′-TCA TAC GAC TCA CTC TAG GG-3′
M2:5′-CCC TAG AGT GAG TCG TAT GA-3′
C3:5′-ACT GAC TGA CTG ACT GAC TG-3′
M3:5′-CAG TCA GTC AGT CAG TCA GT-3′
具有安装在5′-末端的巯基的寡核苷酸得自Integrated DNA Technologies(Coralville,IA)。将样品(80μL中2mg)与320μL 10mM Tris(2-羧乙基)膦(TCEP)以及400μL1×TE缓冲液(加入具有1mM EDTA的10mM Tris,用HCl调节pH至7.5)混合并在-20℃冷冻储存直至使用。NHS-PEO
通过将融化的5′-巯基ssDNA(30μL,0.39mM)溶液通过NAP-5尺寸排阻色谱(GEHealthcare)实现DNA修饰。然后,在室温下将洗脱液暴露于20μL NHS-PEO
为了确认修饰化学的性质,制备并表征了寡核苷酸缀合物的模型。要进行制备和表征,用6-氨基-N-(4-氨基苯乙基)己酰胺使0.5mL DMF饱和并加入到NAP-5纯化后所得的1mL反应溶液中。在室温下孵育30分钟后,使用MALDI-TOF MS分析寡核苷酸缀合物。所观察到的质量在期望值的0.090%内。
如下所述进行了活细胞的修饰和链接的DNA分子的定量:在临修饰之前用PBS缓冲液将5×10
为了对表面DNA分子的数目进行定量,将经修饰细胞的一部分在0℃下与10μLFITC标记的互补DNA链溶液一起孵育30分钟。然后,在分析之前,用PBS溶液清洗细胞并在含有1%FBS的PBS中再悬浮。然后,通过流式细胞术分析细胞。使用已知荧光团密度的荧光珠校准荧光测量。将荧光测量值与缺少DNA修饰的对照细胞以及与不匹配DNA序列反应的细胞所对应的那些进行比较。
图31显示了通过上述方法制备的DNA-细胞缀合物,它与用互补DNA链修饰的基底表面结合,这证明了DNA-细胞缀合物的形成。
通过下列程序,使用EDC偶联化学将DNA缀合至细胞:
1.取500万个细胞并用10mL PBS清洗两次。
2.将0.366g EDC溶解在19.06mL PBS中。
3.将0.452g NHS加入到EDC溶液中。
4.确保pH在7.0左右(通常它们是如此)
5.取1mL该EDC/NHS溶液,其中加入了200μL 80μM的胺-ssDNA(在1×SSC中)。
6.在室温下孵育30分钟至1小时。
7.用10mL 1%的FBS/PBS清洗三次。
8.再悬浮到50至100μL 1%的FBS/PBS中,并准备应用于载玻片。
具有细胞壁的细胞上表面暴露的二醇(脂多糖、脂磷壁酸等)的氧化。使用离心作用,在1mL Dulbecco’s磷酸盐缓冲盐水(DPBS)中漂洗细胞(10
酰肼修饰的ssDNA与氧化的表面暴露的二醇的连接。将细胞在800μL pH6的MOPS缓冲液(0.5M NaCl,0.1M 3-(N-吗啉代)丙烷磺酸,18.2MΩ的H
图32显示了通过上述方法制备的DNA-细胞缀合物,它与用互补DNA链修饰的基底表面结合,这证明了DNA-细胞缀合物的形成。
DNA链与细胞连接以及它们在DNA印制表面上固定的一般方案如下所示:在临修饰之前用PBS缓冲液将5×10
要印制玻璃表面,将20μM 5'胺官能化的ssDNA在3×柠檬酸钠缓冲盐溶液(SSC:45mM柠檬酸钠,450mM NaCl,pH7.0)中的溶液用于样品制备。通过手动吸取或通过使用UCBerkeley官能化基因组实验室的机器人微阵列印制系统将DNA溶液沉积到醛官能化的载玻片(SCHOTT Nexterion,Louisville,KY)上。根据生产商的方案,将点样DNA固定化并使载玻片钝化。印制后,将载玻片在N
使用光刻法结合铝剥离技术实现载玻片的微图案化,并且将在其他部分中对其进行完全详细地描述。对于使用一种细胞类型的研究,用C2序列标记所有细胞。除非另作说明,用互补DNA序列M2使载玻片图案化。将DNA修饰细胞的溶液引入到每个表面上并在不加搅拌的情况下孵育3-5分钟。然后,用含有1%FBS的PBS清洗装置两次。在清洗前,通过随机选择三个装置区域采集重复数据集。对每个位置照相、清洗,然后再次显像。
如下所述进行细胞存活的评价:将C2链包被的Jurkat细胞接种到具有正常生长培养基的1mL佩特里培养皿中,并且将M2链DNA加入到2μM浓度的溶液中。作为对照,将未修饰的Jurkat细胞在相同条件下培养。使用血细胞计数仪,在24、48和72小时对四个样品中的每一个进行细胞计数。通过加入台盼蓝监测细胞存活。
通过膜联蛋白V/碘化丙啶染色(BD Biosciences)确定表面结合的细胞的细胞凋亡程度。通过DNA在载玻片上固定后,将细胞在正常培养基中在37℃孵育24小时。作为对照,在相同条件下孵育未结合的Jurkat细胞样品(缺少表面DNA链)。制备了由900μL 1×结合缓冲液、30μL膜联蛋白V-FITC储液和30μL PI储液组成的溶液。24小时后,在室温下将100μL的该溶液施加到载玻片上15分钟。1小时后,通过荧光显微镜使细胞成像并对非活细胞计数。
如下所述实施了粘附细胞系在图案化表面上的固定:从ATCC获得了两个乳腺癌细胞系MCF-7和MDA-MB-231。用1mM不含任何胰蛋白酶的EDTA将细胞从培养板上去粘附,用PBS清洗细胞溶液三次。将5mL的细胞溶液部分(1×10
如下所述确认了细胞固定化的序列特异性:如上所述,通过将每个细胞群体与NHS-DNA(分别为序列C2或C1)在PBS中孵育30分钟制备了DNA修饰的Jurkat细胞和MDA细胞。为了有助于细胞的目视区分,用CellTracker BlueTM或CellTracker GreenTM活细胞染色标记了每个群体的胞质溶胶。漂洗后,将等量的每个群体混合,引入到具有序列M2或M1(如上文所构建的)的微点样DNA微阵列上,并孵育5分钟。然后,用含有1%FBS的PBS清洗微阵列两次并在荧光显微镜下观察。
如下所述完成了人红细胞的固定:新鲜的红细胞样品得自健康人的血样并且在室温下保存在1%的柠檬酸溶液中。细胞在1小时内使用。用PBS清洗细胞溶液三次,然后在NHS-ssDNA溶液中孵育30分钟以允许细胞表面的修饰。然后,在包被到具有互补ssDNA链的载玻片上之前,用1%FBS/PBS溶液清洗细胞悬液三次。细胞连接后,用1%FBS/PBS清洗载玻片以除去任何未结合的细胞并在光学显微镜下查看。固定后,在1%FBS/PBS中孵育细胞,并且在3小时后使用台盼蓝染色检查它们的存活。
如下所述进行了原代CD4+T细胞的图案化和IL-2产生测定:原代CD4+T细胞(通过与Jay T.Groves实验室(UC Berkeley)的合作获得)收获自小鼠并且在使用之前在报道的条件下生长。然后,使用NHS-DNA方案修饰原代T细胞并将其暴露于通过如上所述的点样或使用光刻法印制的不同DNA图案。用1%FBS/PBS清洗具有DNA-固定的细胞的载玻片以除去任何未结合的细胞并在光学显微镜下查看。
使用ELISA检查了用DNA双链体固定的原代T细胞的IL-2产生。用DNA链修饰了2×10
如下所述进行了原代成肌细胞图案化:从小鼠中收获了原代成肌细胞(与RandallLee实验室(UCSF)合作获得)并根据Huang,N.F.;Patel,S.;Thakar,R.G.;Wu,J.;Hsiao,B.S.;Chu,B.;Lee,R.J.;Li,S.Nano Lett.2006,6,537-542(作为参考并入本文)中所发表的方案进行纯化。简要地,在加入10%(v/v)胎牛血清(FBS,HyClone)、1%bGF(Invitrogen)和1%青霉素/链霉素(P/S,Sigma)的汉姆氏F-10培养基(Ham’s F-10media,Invitrogen)中实现了正常细胞生长。在临表面修饰前用不含任何胰蛋白酶的1mM EDTA使细胞去粘附。用PBS漂洗所得细胞悬液三次。将5mL的细胞溶液部分(1×10
开发了在pH敏感微电极阵列上进行DNA条形码靶向的单细胞捕获的微装置以用于代谢分析。用膜结合单链DNA修饰细胞,并且通过在微流体通道内氧化铱pH微电极传感器区中结合的互补链指导特异性单细胞捕获。显示该双官能微电极阵列能够用于pH监测以及原代T细胞和Jurkat T淋巴瘤细胞分化。单个Jurkat细胞显示出11毫pH/分钟的细胞外酸化速率,而原代T细胞仅表现为2毫pH/分钟。该系统可用于特异性捕获非粘附细胞,并且根据它们改变的代谢特性来区分异质集合体中视觉上相似的健康和癌性细胞。本发明所示的双官能微电极阵列显示使用本发明组合物所制备的装置如何具有选择性捕获细胞和测量其电学和代谢活性的能力。使用DNA-条形码捕获,可以在同一装置上研究粘附和天然非粘附细胞。另外,这样捕获的细胞不是活化的。阵列形式允许直接区分混合物与细胞,从而显示了单细胞性能的变化和细胞如何对整体产生贡献。受控单细胞电化学测量表明使用纳米尺度的细胞界面来进行多重亚细胞的细胞活性分析。
对开发用于单细胞分析的集成微装置来说,微流体装置中单细胞的受控捕获是必不可少的。通过使用与个体细胞相当的尺寸和体积尺度,微流体装置提供了控制细胞微环境的有力工具。已表明在DNA捕获细胞可以用于执行微流体芯片中的单细胞基因表达分析并且用该基因分析来代表未活化的细胞之前,工程设计的细胞表面DNA(细胞粘附条形码)的使用能够捕获细胞。本发明所述的装置具有使用DNA条形码细胞捕获来布置具有未活化(天然)细胞的pH敏感微电极阵列的新能力。该系统使得能够在pH传感器表面上进行快速、选择性和可逆的非粘附单细胞(先前是不可能的)以及粘附细胞的捕获。基于细胞外酸化与总能量使用成正比的原理,这种双官能系统使得能够精确地对单细胞代谢进行实时监测。在这些实验中,已证实该技术可鉴定具有高代谢活性的癌细胞,从而使该装置成为癌症治疗的潜在诊断和预后工具。另外,由于该系统可经过调整从而对所研究的细胞具有极高的选择性和特异性,并且这些细胞可置于装置表面上的精确位置处,因此对于任何疾病(当然,包括癌症),该系统对被称为患者特异性分析和治疗的应用来说是理想的。
先前的工作已表明了微流体系统中单细胞捕获和pH值监测的各个方面。已显示了排成阵列的单细胞捕获的多种方法,其包括物理和能量阱,以及生物化学粘附。该先前工作中的细胞不是使用DNA捕获的,而是通过装置环境中的屏障物理性限制的。尽管简单的限制性捕获孔和微流体陷阱可用于在传感器上分离细胞,但是已表明对于正常细胞功能来说,对新鲜培养基的接近以及清除废物的能力是重要的。如果将要使用亚细胞级电极,那么对于监测活动来说,高度精确的细胞布置也是重要的。本发明的寡核苷酸连接系统使得能够不顾装置上的物理屏障而进行捕获,并且使得能够在该过程中在没有细胞损失风险的情况下对细胞进行补给和清洗。
在细胞活性的定量分析中,细胞外酸化的使用是有价值的工具。重要实例是Cytosensor Microphysiometer,它已作为定量代谢的方法广泛用于测量整体细胞群体(10
本发明所述的装置提供了基于寡核苷酸的细胞捕获技术与尺寸尺度和个体细胞相同的传感器的直接集成,因此允许进行之前在双官能电极系统中从未实现的细胞分析。将通过光刻图案化的氧化铱pH微电极阵列封闭在微流体通道内。使用硅烷接头将单链DNA连接到氧化铱表面上,从而使传感器具有捕获具有互补DNA的细胞的能力并同时保留其检测灵敏度。在本发明中,我们使用该系统测量由非粘附T细胞的代谢所造成的细胞外酸化,并且我们证明了pH灵敏度足以将健康的原代T细胞与具有较高代谢的癌性Jurkat T细胞区分开。我们的结果显示了相同基本类型的个体健康的和转化的细胞的差异代谢活动,这可以使得能够在异质样品内鉴定循环中肿瘤细胞(circulating tumor cell,CTC)。DNA引导的细胞捕获与双官能电极上电化学监测的新型组合提供了用于单细胞分析的新平台。
如下所述完成电极传感器的制造:如前所述,使用标准光刻剥离,在1.1mm厚的硼硅浮法玻璃板(borofloat glass wafer)上使电极(具有20nm Cr粘附层的40nm厚的金)图案化(图11)。使用Specialty Coating System Labcoter 2Parylene(聚对二甲苯)沉积系统,将7μm厚的Parylene-c层沉积到板上,并且使用AlphaStep IQ轮廓测定器进行测量。将100nm的铝层蒸发到装置上,然后在60℃使用具有表面活性剂的Air Products铝蚀刻剂光刻蚀刻30s(图11A、11B)。铝层的蚀刻掩模是通过光刻图案化的1μm厚的Shipley 1818光致抗蚀剂薄膜。然后,使用氧等离子(60sccm O
从传感器区除去聚对二甲苯隔离物后,按照Yamanaka等人的方案,用氧化铱层电沉积传感器。简要地,如下制备了铱沉积溶液。将37.5g IrCl
沉积后,将该装置进行1分钟的等离子体清洗并通过在60℃汽相淀积60分钟用三甲氧基甲硅烷基丙醛进行修饰。然后,将胺修饰的ssDNA(80μM的磷酸盐缓冲盐溶液)沉积到该装置上并使用如前所述的还原胺化进行结合(图11C)。DNA沉积后,通过在室温下经搅拌用0.1M NaOH处理20分钟溶解了保护性铝层,从而在传感器表面上仅留下捕获DNA(图11D)。
如下所述完成微流体装置的制备:使用Dow Corning Sylgard184,用SU-8或聚苯乙烯模具制备了聚(二甲基硅氧烷)(PDMS)通道。通道为5mm宽,15mm长和600μm高。使用18号钝头针打孔得到与20号聚四氟乙烯管相适应的流体入口,并在另一端打孔得到5mm直径出口储罐。用UV/臭氧系统清洗PDMS通道10分钟,并将其应用于该装置。用DI水充满通道1h以使得氧化铱层水合,然后使用标准pH4、5、7和10缓冲液对电极的pH响应进行校准。使用具有MinCO聚酰亚胺加热器和Cole-Parmer DigiSense PID温度控制器的加热铝制平台将通道维持在37℃。
如下所述完成细胞制备和标记:在加入10%胎牛血清(FBS)和1%青霉素-链霉素溶液的RPMI-1640培养基中培养Jurkat细胞。将培养的细胞维持在37℃和5%CO
如上所述,使用共价修饰细胞表面上伯胺的NHS-DNA缀合物实现了使用ssDNA的细胞表面标记(图11E)。简要地,将细胞在室温下在120μM NHS-DNA在PBS中的溶液中孵育30分钟,然后清洗三次以除去任何未结合的DNA。使用先前报道的点样DNA微阵列载玻片测试了条形码特异性细胞捕获(参见Douglas等人,Lab Chip,2007,7,1442-1448)。
如下所述完成代谢监测:将细胞以10
记录了氧化铱电极和来自World Precision Instruments的远端FLEXREF Ag/AgCl参比电极之间的电压测量值。细胞区域外的相同氧化铱电极用于补偿任何传感器漂移。将传感器电极连接到具有16位模拟向数字转化的National Instruments PCI-6031E数据采集卡上。使用常用的Labview VI,以3Hz多重采样监测数字化信号。使用Peak Fit软件,以1%Loess滤波器处理电压信号以降低噪音。
在代谢分析前,使用标准pH缓冲液鉴定电极(图12)。发现这些DNA-修饰的电极保留了它们的pH灵敏度,其性能与未修饰的氧化铱传感器相当。电极响应稳定并且快速,其在500ms内对1个pH单位的变化产生响应。电极的pH响应通常为-68.5mV/pH单位,在pH范围4至10内线性响应。细胞酸化测量的典型范围在约6.5至7.5,因此该传感器非常适合于这类测量。所观察到的响应幅度与先前所证实的其他水合氧化铱传感器的-60至-80mV/pH的范围一致。Olthuis等人已描述了提供pH灵敏度的电极反应。-60至-80mV/pH的灵敏度范围取决于通过多种电化学技术沉积的氧化铱薄膜的氧化状态。Olthuis提供了提供灵敏度的电极反应。
2Ir(OH)
亲和捕获DNA探针与双官能微电极阵列芯片上的pH微电极的集成为直接监测细胞的细胞外酸化提供了平台,所述细胞通常为非粘附的。该系统还为未通过寡核苷酸连接系统活化的细胞(无论是粘附还是非粘附细胞)提供了监测。如图13中所示,尺寸限制的双官能微电极使得能够直接在传感器上进行单细胞捕获。通过测量Jurkat和原代T细胞的细胞外酸化测试了双官能微电极阵列。首先,在阵列上分开地捕获并监测Jurkat和原代T细胞以建立传感器功能性以及两种细胞类型之间的单细胞酸化差异。图14A显示了10分钟的时间段内单细胞酸化的数据。Jurkat细胞显示出11.5±3.3毫pH/分钟的细胞外酸化速率,而原代T细胞表现为1.61±1.5毫pH/分钟(s.d.,每个的n=9)。还用整体细胞群体酸化测量确认了这种差异(在37℃,在低缓冲培养基中的约10
为了证明区分混合群体中不同细胞的能力,在阵列上同时监测了来自具有相同细胞粘附条形码的Jurkat和原代T细胞的混合物中的单个细胞。图14B显示了阵列上10分钟内混合细胞的酸化数据。所测量的酸化速率的差异与分开的样品的趋势相同,并且它允许在两种目视相似的细胞之间进行区分(图14B)。Jurkat细胞的酸化速率为10.1±2.3毫pH/分钟,而健康T细胞为2.41±2.54毫pH/分钟(s.d.,每个的n=5)。
图14C显示了在阵列中使用已知细胞群体的几次试验中的酸化速率的柱状图。对于Jurkat细胞,平均酸化速率为11.5±3.2毫pH/分钟,而原代T细胞显示出的速率为1.62±1.31毫pH/分钟。该差异明显是显著的,其中T检验值p<0.0002。尽管Jurkat细胞略大于原代T细胞(通常直径为12μm相对于10μm),但是尺寸差异不足以解释酸化差异。
为了证明测量单细胞对外源刺激响应的能力,用鱼藤酮处理Jurkat细胞并在双官能微电极阵列上捕获(图15)。预计用鱼藤酮孵育将干扰线粒体电子传递链,从而导致细胞转变为乳酸发酵以完成糖酵解循环。那么,所得的乳酸分泌应提高细胞环境中的酸化速率。在实验中,首先在正常条件下孵育捕获的细胞以建立需氧代谢下的基线酸化速率(约8.8毫pH/分钟)。13分钟后,将10μM鱼藤酮加入到通道中,其导致在1分钟内酸化速率提高3倍(约27.7毫pH/分钟)。其中用1μM鱼藤酮的低缓冲培养基(约10
本发明所开发的双官能微电极阵列在阵列形式中将选择性细胞捕获和单细胞代谢监测这两种重要功能相结合。在早期的工作中,Castellarnau等人使用双向电泳定位ISFET pH传感器附近的细菌高浓度悬液并测量了葡萄糖存在下细胞的酸化。尽管这种技术非常适合于测量整体响应,但是它缺少分辨单一细胞独特活性的能力。Ges等人的单一心脏细胞pH系统提供了监测大的粘附细胞的能力,但是密封通道所造成的体积排除使其难以引导细胞连接。DNA条形码捕获提供了引导捕获粘附和天然非粘附细胞(如T和B细胞)的优势,以及分析未被寡核苷酸连接方法所活化的细胞的额外优势。这种受控捕获为细胞表面上活力的空间分辨的电学和/或光学探测和测量提供了平台。
酸化数据显示,在用捕获DNA处理并连接至电极后,单个非粘附细胞继续正常表现。尽管任何捕获技术都可能对细胞产生一些影响,但是细胞粘附条形码绕过了通常基于整合素或抗体的捕获所用的天然细胞表面受体,并因此避免了那些已知信号途径的激活。对于Jurkat和原代T细胞来说,所测量的细胞外酸化速率与Ges等人所报道的单个细胞酸化速率相当,而我们的官能化微电极技术灵敏度的提高使得能够区分这两种细胞类型。
我们的单细胞结果显示可以在单细胞水平上检测原代非转化细胞和永生化癌性T细胞的代谢活性之间的差异。我们已证明了使用此代谢差异对相同基本类型的目视相似的单个细胞进行电化学区分的能力。该方法可用于通过其独特代谢活性来鉴定个体循环肿瘤细胞,优于简单的基于抗体的捕获。它还可用于区分不同转移潜能的癌性细胞。这种混合物中的单个细胞监测允许基于癌症发展或起源的细胞状态来检测药物响应的差异。
所述具有明显扩展性以包含更多元件的阵列形式允许直接比较相同条件下多种细胞的个体活性,并具有足够的能力来鉴定整体变化。纳米制造的电极阵列的构造可产生具有高空间分辨率的细胞表面电化学分析谱图。先前已使用扫描电化学显微镜显示了静态细胞表面轮廓,但是纳米电极阵列可以将其从连续过程转化为并行过程并且还提供了活细胞中的时间分辨率。
该系统能够提高来自单个细胞的检测分析物的数目,并且一般具有应对分析系统复杂度提高的能力。对先前所提到的用于整体细胞监测的Cytosensor Microphysiometer系统进行了改造,使得除了标准pH测量能力之外,还同时测量葡萄糖、乳酸和氧水平。还可以将微米或纳米制造的分析物选择性传感器添加到该系统以提供额外的分析深度,其包括单个细胞上的多分析物传感。Thayer等人已将钙敏荧光团和电控制的组合用于监测膜片箝记录期间单个神经元中的钙通量(calcium flux)。容易地改造PDMS/玻璃多层装置以使得能够同时进行荧光和电测量。尽管荧光探针经常受到光漂白的影响,但是本发明所述技术可用于在几小时或几天内追踪单个细胞的代谢活性,从而揭示细胞在其生命周期过程中的任何变化。
基于DNA条形码的细胞捕获提供了设计个体细胞间连接的能力,它使得能够构建和分析电极上离散的多类型细胞系统。例如,可以使用DNA将单个神经元与单个肌细胞相连,以允许分析单个细胞神经肌肉突触的形成和作用。另外,可使用通过寡核苷酸杂交的细胞与细胞的连接来设计人工组织。可以将传感器置于系统中或附近以记录系统中的代谢、电学或其他变化。
单细胞分析是理解等基因细胞群体内基因表达变化的有力方法。例如微阵列和基因表达系列分析的常规基因表达分析技术不够灵敏,无法分析单细胞水平的变化,仅报告大量细胞的整体平均表现。最近,已开发了多种高灵敏度的专门技术来探测单个细胞中的基因表达。尽管多种这些方法提供了实时监测的优势,但其方案需要大量劳动,通常需要细胞工程学并且多重能力是有限的。
新近开发的微流体技术和方法使得能够以可以放大到大量细胞的形式进行单细胞分析。微流体装置提供了探测单个细胞的强大平台,这是因为其固有长度(1-100μm)和体积的尺度(皮升至纳升)与单个细胞的尺寸和体积(约1pL)接近。微流体所提供的最大优势在于将所有处理步骤集成到单个装置中的能力,消除了将会阻碍灵敏且可重复的定量单细胞分析的样品污染和产物损失。
为进行单细胞基因表达分析必须集成到微装置中的3个步骤为细胞选择和定位、酶促反应以及目的分析物的定量检测。尽管多种微流体系统已显示了这些要素中的1个或2个,但是成功集成所有3个要素是极其困难的。早期微流体系统成功地将PCR室与毛细管电泳(CE)分离通道相结合。最近的集成微系统已表现出检测灵敏度的显著提高、粗样品的操纵和大规模并行性。除了这些发展之外,尚没有集成微流体装置能够成功地将所有3个步骤结合到一个平台中来直接测定单个细胞基因表达的变化。根本的障碍在于每个纳升处理步骤之间分析物的有效转移。
为了解决该困难,我们已开发了具有单细胞基因表达谱分析的所有必需要素的集成微流体装置,并且使用该装置进行了单个细胞基因沉默的研究(图20)。细胞表面上20个碱基的寡核苷酸对细胞官能化使得能够通过DNA杂交在金垫上进行捕获。对2个细胞群体进行了GAPDH mRNA和对照18S rRNA的多重基因表达分析。第一个细胞群体是由在正常条件下生长的未处理的Jurkat T淋巴细胞组成,其表现出两种靶基因的均匀高表达(图20A)。用针对GAPDH mRNA的siRNA处理第二个细胞群体(图20B)。相对于18S rRNA对照,探测了个体细胞中GAPDH mRNA的沉默程度。
该基因表达微装置在100mm直径的玻璃晶片上包含了4个独立可寻址的阵列分析系统(图21)。每个相同的微系统包含4个独立的区域,它们集成在一起以实现处理步骤之间的最大转移效率。第一个区域是用于将材料从样品入口移动通过反应器区的3阀泵。在反应器区中,使单个细胞捕获、裂解,并且通过RT-PCR将所目的mRNA逆转录并扩增。亲和捕获区包含用作储罐的容留室和在亲和捕获凝胶基质中固定、纯化和浓缩扩增子的捕获室。最后,该系统包含用于基于尺寸的分离和产物定量的CE分离通道。
从单细胞捕获到CE分离和检测的全部分析是在<75分钟内进行的,如图22A中所示。首先,用20个碱基的寡核苷酸使Jurkat细胞官能化(图22B)。用被细胞代谢并导致在细胞表面上出现叠氮基团的合成过乙酰化N-叠氮乙酰基甘露糖胺(Ac
在另一个实施方式中,可以使用如实施例1中所述的本发明方法将ssDNA分子连接到Jurkat细胞上,如下所示:在临修饰之前用PBS缓冲液将5×10
在反应器内部,通过金-巯基连接将巯基修饰的捕获DNA的互补的20个碱基的链固定在光刻限定的25×25μm
将残留的未捕获细胞清洗出反应器后,制备了用于分析的捕获的细胞(图22C)。在30s快速冻融裂解后,在42℃下孵育15分钟的时间内,将靶标mRNA逆转录为稳定的cDNA链。然后,在相同的200nL反应器中在25分钟内完成30个PCR扩增循环。然后,定量地将所有RT-PCR产物从该反应器中转移到用于尺寸分析的分离通道。将扩增的片段和未反应的RT-PCR混合物从反应器泵至容留室中,并且通过电泳从废料罐驱动到阴极罐中。将对亲和捕获探针具有互补性的目的片段定量地浓缩并固定在捕获室的入口,从而产生了纯化的捕获栓(capture plug)。亲和捕获基质包含线性聚丙烯酰胺(LPA)凝胶,其与对目的片段互补的两种20碱基寡核苷酸捕获探针(20μM)共聚合。该捕获过程使用了序列特异性螺旋侵入以固定dsDNA扩增子。捕获探针分别与GAPDH和18S rRNA扩增子末端的23和47个碱基的序列互补。通过将探针置于启动位点内部,捕获凝胶还起到了纯化基质的作用以除去未反应的高摩尔浓度的FAM标记的引物。最后,将纯化和浓缩的产物在80℃下从亲和捕获凝胶上热释放,电泳分离,并通过共聚焦荧光检测定量(数据未显示)。
集成微流体基因表达分析系统获得了个体细胞基因沉默的定量数据。如所预期的,未处理的Jurkat细胞显示出正常的GAPDH mRNA和18S rRNA表达(图23A)。代表性电泳图谱显示了分别对于200bp GAPDH和247bp 18S rRNA靶标,在160s和185s处迁移的两个强峰(图23)。此外,使用捕获基质来固定目的片段使得从分离中移除了所有未反应的引物,从而使我们能够无干扰地观察小的和大的扩增子。用靶向GAPDH mRNA的siRNA电穿孔的单个Jurkat细胞仅在185s产生了18S rRNA的单个峰。8个个体细胞的单细胞实验显示GAPDHmRNA的表达是未处理的Jurkat细胞的0、5、50、1、48、0、5和0%(图23B)。该分析表明将单个细胞归为中度(约50%)或完全沉默(约0%)的群体。这些单细胞测量从根本上不同于在相同条件下对50个Jurkat细胞进行的整体测量,其中GAPDH的表达降低至其原始值的21±4%(n=4)。因此,对基因沉默所测量的整体平均值掩盖了个体细胞响应的随机多样性。垫上无细胞捕获的对照测定显示无产物,从而证明系统中无残留污染。类似地,无逆转录酶的PCR对照显示无扩增,从而确保扩增模板是RNA而非DNA。
为了确保沉默行为的变化不是电穿孔过程中所引入的siRNA量的简单函数,用荧光标记的siRNA处理细胞。在标准条件下生长的细胞显示Cy3-标记的siRNA的平均摄取为17±2相对荧光单位(relative fluorescence unit,rfu)(表S1),其中在30个细胞中有4个表现出22rfu的略高的水平。如果电穿孔的变化是两个细胞群体的原因,那么我们预期将有13%的细胞被鉴定为完全沉默而87%为中度沉默。在微流体装置中进行的单细胞基因表达分析显示出相反的趋势(图23B)。
表S1,单细胞siRNA电穿孔实验的列表数据
用Cy3标记的siRNA对细胞进行电穿孔,在正常条件下培养并进行荧光成像。对每个细胞进行询问,记录最大像素强度值。另外,记录刚刚细胞外的最小值用以归一化。平均摄取为17.2±2.4相对频率单位(rfu)。最大摄取为23rfu,最小为15rfu。
另外,对GAPDH基因测序以检查细胞群体经受的中度沉默是否是由siRNA结合域中的杂合多态性所引起的。发现该序列没有突变。GAPDH siRNA结合序列的测序结果显示在Jurkat细胞中存在预期的序列(5'-AAA GTT GTC ATG GAT GAC C-3'),这表明两种细胞群体不是由结合域中的多态性所引起的。因此,本发明所显示的这两种细胞群体不是siRNA递送、遗传变异或细胞存活所致的普通结果。
在Liu YP,Dambaeva SV,Dovzhenko OV,Garthwaite MA,Golos TG.Stableplasmid-based siRNA silencing of gene expression of human embryonic stem cellStem.Stem Cells Dev.2005;14:487-492中已检测到具有不同基因沉默水平的两种不同细胞群体的出现。人胚胎干细胞(hESC)中绿色荧光蛋白产生提高的测量显示出对siRNA处理或全或无的响应,但是对其潜在机制尚未鉴定。我们的系统显示了进行定量转录本分析的独特能力,从而揭示尽管80%的细胞显示出预期的完全抑制,但是20%显示出50%的抑制。这表明遗传或表型的双稳定性或开关的存在,其控制siRNA降解、阻断其靶标结合或抑制转录本降解。在这些中,仅有后两种机制能够提供对50%抑制水平的有效解释。现在,需要更详尽的研究来验证本发明中所观察到的双相表达趋势和研究其机制起源。由于高度并行的结构的制造是我们的方法的重要优势之一,因此放大至96个分析器将提供这类研究所需的通量。
在我们的表达微装置中通过单个反应器中仅30个PCR循环来实施一步RT-PCR扩增的能力是有效集成的直接结果。首先,使用玻璃而不是多孔聚合物材料防止产物吸收。第二,玻璃的高导热率使得能够快速进行热循环并提高反应效率。第三,气动阀和泵的使用使得能够有效地转移纳升材料团块。最后,亲和捕获、纯化和浓缩过程使得能够对所有产生的产物进行定量分析,这是相对于使用常规交叉注射器(cross injector)或水动力压力注射器(hydrodynamic pressure injector)的显著改善,它仅允许对小部分(<1%)的产物进行分析。我们的集成装置的这些有利属性为单个细胞性能和行为的多种生物分析研究指出了方向。
在本文中,我们已经对由于siRNA处理所造成的mRNA敲低(knockdown)的变异性进行了单细胞测量。该测定显示出被整体测量所掩盖的单个细胞中独特的双相基因抑制效率。由于该分析步骤使用了基于尺寸的分离,因此可以产生和分析的产物数目决定了多重能力,表明可以平行地研究5-10个靶标的表达。通过将该微装置与激光捕获显微解剖相结合,可以在单细胞水平上研究肿瘤的异质性质。当使用Oct4 mRNA靶向来引发向滋养层样细胞的分化时,还有可能进行针对siRNA处理对hESC中表达影响的定量单细胞分析。此外,根据我们先前对<11个mRNA分子/反应器的检测,一旦完全实现并集成改善的产物捕获、纯化和注射过程,则我们的微流体装置最终可以使得能够在单个转录本水平上研究个体细胞的表达。总的来说,我们的方法为揭示隐藏在整体平均值之下的基因表达随机变化提供了多种令人激动的前景。
材料和方法。生物处理器制造。制造方案与之前的核酸扩增微装置Toriello NM,Liu CN,Mathies RA.Multichannel reverse transcription-polymerase chainreaction microdevice for rapid gene expression and biomarker analysis.AnalChem.2006;78:7997-8003中所使用的类似,该参考文献作为参考并入本文。简要地,要形成气动系统歧管晶片,在0.5mm厚的100mm的硼硅浮法玻璃晶片上光刻限定阀座(valve seat)和动作通道(actuation channel)并蚀刻至38μm深。钻孔得到阀动作出入孔,并将歧管切成可重复使用的9mm×6cm的条。通过用UV臭氧清洗剂活化254μm PDMS膜的两侧1.5分钟以改善PDMS-玻璃的粘结,然后将该膜夹在歧管和粘合的通道晶片之间从而形成了可拆卸的聚二甲基硅氧烷(PDMS)弹性体阀。
在0.5mm厚的硼硅浮法玻璃晶片上制造了反应器/通道晶片。在正面光刻限定了用于泵送的流体通道并蚀刻至38μm深。在背面光刻限定了反应室、容留室和捕获室以及分离通道并将其蚀刻至20μm深。用金刚石钻出电泳储罐、电阻式温度检测(resistancetemperature detection,RTD)出入孔和阀通孔。为了形成RTD板,对用200埃的Ti和2000埃的Pt(Ti/Pt;UHV溅射)溅射沉积的0.5mm厚的硼硅浮法玻璃晶片光刻图案化并用90℃的王水蚀刻形成30μm宽的RTD元件和300μm宽的导线。将钻孔的反应器/通道晶片对齐并通过使用可编程真空炉在655℃加热6小时以热粘合至RTD板。
为了形成可拆卸的模块加热器,用2200埃的Ti/Pt溅射沉积0.5mm厚的硼硅浮法玻璃板。通过将6μm的金电镀到光刻限定的区域上形成了加热器导线。通过在离子磨中各向异性地蚀刻光刻暴露的Ti/Pt形成了连接金导线的Ti/Pt蛇形电阻加热器元件。
Jurkat细胞制备。将T淋巴细胞Jurkat细胞在50mL培养瓶(Nalge-NuncInternational)中在含有1%青霉素/链霉素(1%P/S;Invitrogen)和25μM Ac
siRNA处理。对于基因沉默研究,用2.5μg双链GAPDH siRNA(有义,5'-GGU CAU CCAUGA CAA CUU UdTdT-3';Ambion)对150000个Jurkat细胞电穿孔。将细胞在75μL的siPORT电穿孔缓冲液(AM1629;Ambion)中悬浮,并且在1mm比色杯(Bio-Rad)中以250V进行250μs的单次脉冲。然后,以与上文Jurkat细胞制备一节中所述相同的方式培养并准备细胞。对于阴性对照研究,用不结合mRNA的150pmol的Cy3标记的siRNA对细胞电穿孔。
RT-PCR混合物。对直接来自Jurkat细胞的GAPDH和18SrRNA转录本进行多重RNART-PCR。25μL RT反应混合物包含Cell-to-cDNA II试剂盒[4单位的莫洛尼鼠白血病病毒(Mo-MLV)逆转录酶、0.4单位的RNA酶抑制剂、0.1μM的dNTP、1×RT缓冲液(Ambion)]、0.08单位的铂Taq聚合酶(Invitrogen)以及800nM的GAPDH基因正向和反向引物以及20nM的18SrRNA靶标正向和反向引物。GAPDH正向(5'-AGG GCT GCT TTT AAC TCT GG-3')和反向(5'-FAM-TTG ATT TTG GAG GGA TCT CG-3')引物产生了200bp的扩增子。18S rRNA正向(5'-CGGCTA CCA CAT CCA AGG AAG-3')和反向(5'-FAM-CGC TCC CAA GAT CCA ACT AC-3')引物产生了247bp的扩增子。分别通过在反应混合物中去除Mo-MLV RT和Jurkat细胞在微装置上进行了无RT和无模板的对照。
基质合成。通过将LPA与2条5'-acrydite-修饰的捕获寡核苷酸共聚来合成DNA亲和捕获凝胶。在4℃下,通过用氩气鼓入含有6%(w/v)的丙烯酰胺、1×TTE和40nmol的2种acrydite修饰的寡核苷酸(IDT)的2mL溶液2h,然后加入0.015%(wt/vol)的过硫酸铵(APS;Fisher Scientific)和四甲基乙二胺(TEMED;Fisher Scientific)合成了亲和捕获基质。亲和捕获基质含有针对GAPDH的捕获探针(5'-Acry-ATC CCA TCA CCA TCT TCC AG-3',T
反应器制备。通过使用改进的Hjerten涂层方案,用聚二甲基丙烯酰胺(PDMA)使玻璃表面衍生化以防止非特异性细胞粘附。首先,通过与1M NaOH孵育1h将反应器玻璃表面去质子化。用0.6%(vol/vol)的(γ-甲基丙烯氧基丙基)三甲氧基硅烷(γ,Sigma)在3.5pHH
然后,通过与三(2-羧乙基)膦(TCEP,200μM;Invitrogen)去保护的巯基-DNA(5'-巯基-AGT GAC AGC TGG ATC GTT AC-3',20μM)一起孵育1h,用ssDNA使反应室中心的光刻限定的25μm×25μm的金垫官能化。然后,漂洗并干燥该室以除去未结合的DNA。
凝胶上样序列。通过用在甲醇中稀释的动态涂层(1:1;DEH-100;The GelCompany)处理分离通道、容留室和捕获室1分钟以抑制电渗流,从而制备了用于亲和捕获和分离的装置。在室温下,从每个阴极(C)储罐加载多重亲和捕获基质(20μM,6%,黄色)向上至交叉的分离通道。然后,从中心阳极(A)加载分离基质(红色)穿过捕获室至样品装载交叉。在所有阀打开的情况下,通过在样品端口加入3μL无RNA酶的水并在废料(W)罐处施加真空使系统的其余部分水化。然后,将微装置置于44℃的温度控制平台上。用另外2μL的水冲洗系统,以样品装载交叉除去受热膨胀的凝胶。
生物处理器操作。如图22中所示,通过制备用于细胞捕获的反应室开始微装置的操作。将经修饰在其表面上包含ssDNA的细胞悬浮于反应混合物中并通过真空吸入到反应中。当细胞流过细胞捕获金垫时,细胞表面上的ssDNA和固定在金垫上的互补ssDNA之间发生DNA杂交。为了使捕获效率最大化,实施了15分钟的DNA介导细胞捕获。将未捕获的细胞清洗出系统并从废料端口中除去,关闭阀以进行热循环。在该研究期间,所有4个反应器平行使用。这使得能够同时对4个个体细胞进行分析。捕获后,记录每个细胞的形态以确保其仍存活。单细胞研究的总数据采集时间为1个月。
在单个200nL反应器中进行了冻融裂解和一步RT-PCR热循环。将一块干冰放置在所有4个反应器的反应室上30s以使得捕获的Jurkat细胞冻融裂解。在该一步RT-PCR中使用了冻融裂解以防止热启动Tag聚合酶的过早不希望的活化,从而防止逆转录酶变性以及最大程度地降低RNA酶对RNA的降解。然后,在42℃下,通过使用与目的RNA(GAPDH和18S rRNA)转录本互补的引物进行15分钟由细胞RNA合成线性cDNA。在cDNA合成后,使Mo-MLV RT变性,并且在95℃下活化铂Taq聚合酶60s,然后进行95℃,5s;47℃,20s和72℃,25s的30个PCR循环。由于快速的加热和冷却速率(>15℃/s),在50s中完成了每个PCR循环,并且总反应时间为46分钟。
在热循环后,对目的产物进行了亲和捕获、纯化和浓缩。通过使用5步泵循环,将反应器内容物泵入容留室。每步使用350ms的动作,得到30nL的工作容积。在每次泵循环之间使用了23s的延迟,为分析物留出了足够的时间以迁移到捕获区并防止分析物在容留室中累积。废料(W)和阴极(C)罐之间恒定的100-V/cm的电场通过电泳驱动分析物向捕获室运动。在捕获室入口处,与捕获探针互补的分析物发生杂交,产生样品栓。维持废料和阴极之间的电场直至将残留的PCR反应物(过量引物、盐和缓冲液)清洗到阴极罐中,因此产生了纯化的扩增子样品栓。使用30个泵循环,导致总捕获和清洗时间为12.2分钟。在捕获过程完成后,将整个装置的温度升高至80℃以使得捕获的DNA片段从亲和捕获凝胶上热释放,并且用阴极和阳极之间150V/cm的电场分离样品。通过使用激光致荧光和Berkeley旋转共聚焦扫描器(Shi YN等人,Radial capillary array electrophoresis microplate and scannerfor high-performance nucleic acid analysis.Anal Chem.1999;71:5354-5361)检测了所有4个泳道的电泳分离的FAM标记的产物。在扫描器上的温度控制平台上进行整个捕获和释放过程以防止热梯度。
如适用于通过碳水化合物上的醛或酮在非动物细胞上连接寡核苷酸,使用本发明原理可研究并优化产氢效率。寡核苷酸捕获以及与装置的连接使得能够在芯片上产生图案化的细胞和酶。在该系统中,氢气向电的转化测量了氢的产生。由于与传统测量(参见培养顶隙的GC读数)相比对氢产生的潜在较高的灵敏度,通过电流产生来测量直接的氢产生是有利的。本发明对于这些研究和动作的优势包括装置上细胞精确放置所涉及的连接和控制,那么这样燃料电池可以是可重复使用的,并且还包括利用太阳能和碳能的较高效率。例如,使用不同光合细胞的图案化的层,可以构造自持续(self-sustaining)燃料电池装置,其中细胞吸收太阳光谱中非重叠部分的太阳光,并且所述装置还加入了以相邻层中的光合细胞所产生的生物质为生或利用这些生物质的固氮生物的交替的层。当耗尽时,可以加热具有通过寡核苷酸杂交连接的细胞的燃料电池,所述寡核苷酸去杂交,以及清洗掉死细胞。用与装置表面上互补寡核苷酸再连接的新鲜的经修饰细胞漂洗表面。然而,弃去具有包埋了细胞的溶胶凝胶的溶胶凝胶装置,其不能以相同的方式再次使用,但是这种转换对于它们的目标应用可能是适合的。
在37℃下使用在5mM NaIO
所述系统的其他实验和使用将优化芯片上质子交换膜氢燃料电池的设计。另外,必须使用多孔聚合物来防止含氮碱基和细胞废料所造成的铂/钯中毒。为了确定在该系统的置换中的氢输出效率,测量了单位时间所产生的电流。
可以朝着提高系统效率的方向研究不同光合和非光合微生物组合的氢输出效率。可以改变参数例如装置上生物体的图案、光暴露和图案中生物体的特定组合来优化效率。例如,可以采用Melis实验室所提议的三种生物的制氢系统(两种光合自养生物吸收太阳辐射的不同区,而一种异养生物消耗光合作用所产生的生物质并将其转化为光合生物对于自养生长所需的小有机酸)。所提议的生物包括(但不限于),光合:莱茵衣藻(C.reinhardtii)(藻类)、集胞藻属(Synechocystis)PCC6803(蓝细菌)、深红红螺菌(R.rubrum)(革兰氏阴性厌氧菌);异养生物:巴斯德梭菌(C.pasteurianum)(固氮菌)、固氮菌属(Azotobacter)(认为具有任何生物的最高呼吸率)。(参见Melis等人,在本文的其他部分中所引用的)。
在另一个实施方式中,燃料电池装置包含开放空气阴极以代替阴极处基于溶液的还原,和用细菌直接在阳极上图案化的阳极。这将通过用链霉亲和素(通过invitrogen获得)修饰的LPA(线性聚丙烯酰胺)包被阳极来实现。该涂层还将起到保护Pt不受来自细胞的含氮废物的毒害。使用铝剥离光刻图案化,将5'-生物素修饰的DNA在LPA包被的电极上图案化。这样,将首先制造玻璃电极座,然后将DNA在电极上进行图案化,最后细胞将在电极上图案化并且PDMS顶部将粘合在其上方。由于在对数期生长期间并且当营养物丰富时氢产量最大,因此将恒定地使培养基非常缓慢地流入到电池中。将对装置照明并且可以通过(例如)相对于电阻的电压降(对电池设置某种偏差)来测量光合制氢。
在燃料电池装置中,氢气将在Pt电极上成核并且像大多数燃料电池设计一样,将除去电子,从而立即将细胞所产生的氢气转化为电流。
图案化的另一个有趣的方面是使用DNA将细胞在彼此的上方图案化以用于燃料电池装置。现在,参考图30,可以在DNA微阵列上将多个细胞在彼此的上方图案化。除了自发红色荧光的蓝细菌集胞藻(Synechocystis)外,用不同的荧光团标记每个细胞。图30中显示了细胞及其所标记的荧光团的示意图。其优势在于夹在两种光合细胞之间的氧气吸收细胞(棕色固氮菌(A.vinelandii))的空间取向将有可能用于保持光合细胞附近局部的较低氧浓度。由于光合作用产生了作为副产物的氧(至少在藻和蓝细菌的情况下)并且氧抑制了产生氢的酶,因此使光合细胞与具有难以置信的高代谢的细胞相连意味着局部氧浓度应始终较低并且氢的产生是持续的。目前,由于其氢化酶的氧抑制,集胞藻(Synechocystis)在长期H
可以使用气相色谱法测量O
用于修饰每个细胞的链不与其他细胞或玻璃上的任何其他链互补以确保细胞正确地图案化。可以使用在上述实施例中使用的相同寡核苷酸(M1/C1、M2/C2、M3/C3)和两种其他互补序列Z2(5'CACACACACACACACACACA3')和zc2(5'TGTGTGTGTGTGTGTGTGTG3')来使这些细胞在玻璃基底上图案化。这些细胞每次形成一层的图案化。将细胞在5mM高碘酸钠中氧化,然后在约35μM的酰肼-DNA在pH 6的MOPS(N-吗啉代丙烷磺酸)缓冲液(注意,在本实验中所使用的MOPS在其正常缓冲范围之外)中的溶液中孵育。清洗细胞后,将它们与先前用DNA修饰的玻璃基底上的PDMS孔可逆结合。用一种细胞溶液填充孔,以3000rpm离心5分钟,在PBS中漂洗并依次用随后的两种或三种细胞类型重复。如图30所示,在使细胞图案化的这些条件下不应使DNA去杂交。所选择的细胞类型不限于所示的那些,但是使用本发明方法可以添加多个图案化的细胞层。例如,现在参考图30,可以用M2寡核苷酸修饰玻璃。通过C1寡核苷酸,将集胞藻(Synechocystis)细胞层与玻璃基底连接。然后,用C2寡核苷酸修饰该集胞藻(Synechocystis)层。用M2寡核苷酸修饰棕色固氮菌(A.vinelandii)细胞并通过M2/C2杂交将其连接到第一集胞藻(Synechocystis)层。用M2和C3修饰第二棕色固氮菌(A.vinelandii)细胞层,以及用M3修饰第三深红红螺菌(R.rubrum)细胞层,然后将其与M2寡核苷酸杂交并将第三层与第二细胞层相连接。
应在无菌条件下进行图案化,然后将图案放置在所有细胞均可以生长的培养基中和在具有少量顶隙的小瓶中。可以通过随时间从该顶隙中取样并监测活力来监测H
可以使用含有与每种细胞的个体培养基中所必需的营养物浓度相同但是以一种培养基的体积存在的任何培养基。例如,集胞藻(Synechocystis)使用称为BG11的培养基,而棕色固氮菌(A.vinelandii)在伯克氏培养基(Burk’s medium)中生长,因此制备了含有1L BG11和1L伯克氏培养基中所有营养物成分但是以单一体积(1L)存在的共培养培养基,借此提供了与个体培养基相比每升中更高的营养物浓度。深红红螺菌(R.rubrum)使用培养基ORMEROD,因此当所有三种生物的培养将在1L共培养培养基中具有1L每种培养的所有成分(减去用于缓冲的磷酸盐)时,这使得三种培养基处于一种高营养物的培养基中。可以使用伯克氏培养基的磷酸盐缓冲液配方来优化磷酸盐浓度。
该装置将用于恒定地驱动其外部的事物。当细胞死亡时,加热装置,DNA去杂交,清洗掉死细胞,并冲洗入新鲜的细胞以替代老的细胞。该装置还可以用作电池或用作电网的补充。理论上,可以串联多个装置以潜在地驱动车辆或者可以与太阳能硅板具有相同的作用。
对于多种生物过程来说,控制细胞-细胞粘附的作用力是至关重要的,所述生物过程包括细胞分化、组织生长、肿瘤发生以及脊椎动物免疫应答的正确发挥功能。通常通过单个活细胞与能够进行作用力测量的探针(如,微量移液管)连接来表征这些相互作用的强度。最近,已经将光镊应用于捕获单个细胞并用于高精度地测量这些作用力,但是该技术限于施加皮牛顿范围内的作用力。由于能够定量皮牛至纳牛范围的作用力,因此原子力显微镜(AFM)为这些方法提供了引人注意的替代方法,并且该技术的确已用于测量单个活细胞的机械性能和用于研究单细胞水平的粘附力。通过用与细胞表面上的碳水化合物部分结合的纤连蛋白和凝集素包被AFM悬臂,已实现了几种基本粘附测量,但是特别是在后一种情况下,已报道了细胞结合分子本身具有一定程度的细胞毒性,其可影响待评价细胞性质。因此,尽管这些研究突出了AFM对于测量细胞受体-配体相互作用的应用,但是需要一些扩展的悬臂连接方法来研究差别很大的时间尺度内的细胞-细胞相互作用。
为了解决这种需求,我们比较了三种用于将活细胞连接到AFM悬臂上的生物分子介导的方法,其中强调了每种技术可以达到的细胞存活、粘附强度和探针重复使用性。这些研究已表明通过使用互补DNA链的细胞连接对存活的影响最小并且似乎不会激活细胞信号通路。该方法还提供了整体上优良的粘附强度,但是可减弱该参数以允许细胞从一个表面转移到另一个表面。通过采集游离细胞并将它们置于具有更长互补区的DNA链的基底上的精确位置,我们能够证明这一设想。这种“蘸水笔(dip-pen)”活细胞图案化证实了DNA介导的细胞粘附方法的重复使用性并且可以证明对于构建具有良好限定的空间关系的细胞复杂混合物是有用的。
为了允许对几种连接策略进行比较,将三种不同的生物分子(DNA、刀豆蛋白A(ConA)和抗体)连接到用于细胞锚定的氮化硅AFM悬臂上。对于所有连接方法,如图16a中所示,用醛基覆盖工作表面上的氧化硅薄层。通过接触角测量,表征了使用这些步骤所产生的表面。
通过还原胺化,将胺官能化的DNA连接到醛基上(图16b)。首先,将醛包被的悬臂浸没在胺官能化的单链DNA(ssDNA)溶液中,然后加热以促进亚胺形成。冷却至室温后,使用硼氢化钠的水溶液将亚胺还原为不可水解的胺键。该步骤还用于将任何未反应的醛官能团还原为醇。通过在3'末端偶联具有异硫氰酸荧光素(FITC)的5'-胺官能化的DNA链,可以通过荧光成像来验证链的存在。
在上述工作中,将蛋白质通过非特异性吸附连接到AFM尖端上并通过戊二醛交联至引入到尖端表面上的胺基。为了提供更加良好限定的连接(并因此实现更均一的细胞连接),我们选择使用简单还原胺化策略,其用于氨基-DNA链。ConA和抗人CD3抗体(抗CD3)上的表面赖氨酸残基与悬臂表面上的醛官能团反应(图16b),但是使用了较低浓度的还原剂(66μM)以使得二硫键的还原最小化,二硫键是维持蛋白三级结构所需的。使用可商购的样品,容易地实现了在反应中使用的蛋白质浓度(20μM ConA和6μM抗-CD3)。如以上对DNA所述的,在一些实验中使用了FTIC标记的ConA和抗CD3样品以通过使用荧光显微镜验证生物分子连接。对于每种分子检测了相似的荧光水平。
对于DNA、凝集素和抗体与悬臂的共价连接,使用了下列方法。对于DNA介导的细胞粘附研究,设计了互补寡核苷酸序列对(A/A')。序列同一性如下所示:
A:5′-TCA TAC GAC TCA CTC TAG GG-3′
A′:5′-CCC TAG AGT GAG TCG TAT GA-3′
将醛包被的悬臂(MLCT-NONM)在3×盐/柠檬酸钠缓冲液(45mM柠檬酸钠,450mMNaCl,pH7.0)中20μM 5′-胺官能化ssDNA的溶液中浸没15分钟,在烘箱中在100℃加热30分钟,然后用0.2%的SDS溶液和蒸馏水清洗(每次1分钟)。将所得的悬臂在10mL乙醇和30mLPBS溶液中的0.1g NaBH
还通过还原胺化程序将刀豆蛋白A和抗CD3 IgG单克隆抗体偶联到醛包被的悬臂(MLCT-AUNM)表面上。在潮湿室中,将醛包被的悬臂暴露于20μM(ConA)或1mg/mL(抗CD3)的蛋白质在pH7.0的PBS缓冲溶液(含有66μM NaBH
为了制备在其表面上具有ssDNA的活细胞,我们首先将叠氮官能团引入到包埋在质膜中的糖蛋白中,如前所述[Zabzdyr JL,Lillard SJ.Measurement of single-cellgene expression using capillary electrophoresis.Anal Chem.2001;73:5771-5775]。将过乙酰化的N-α-叠氮乙酰基甘露糖胺(Ac
在其他研究中,要制备在它们的表面上具有ssDNA的活细胞,可以实施用于将DNA链连接到细胞上的一般方案。在临修饰之前用PBS缓冲液清洗5×10
使用两种不同的方法评价了粘附分子对细胞存活的影响。第一,向未修饰的Jurkat细胞悬液中添加ConA或抗CD3抗体,并且向DNA包被的细胞溶液中添加互补序列。图17a显示了所得细胞在三天内的生长曲线。DNA修饰细胞的增殖与未修饰细胞的相同,但抗CD3处理的细胞表现出延迟的生长。12h后,ConA包被的细胞聚集并且不再存活。细胞形态发生可溶性生物分子所引起的变化。Jurkat细胞在正常培养基(对照)中生长,DNA修饰的Jurkat细胞在2μM DNA存在下生长,未修饰的Jurkat细胞在2μM ConA或0.1mg/mL抗CD3存在下生长。12h后,用光学显微镜检查所得的细胞。在DNA存在下,细胞外观未发生大地改变,但在ConA和抗CD3存在下生长的细胞表现出聚集和其他形态变化。
作为第二种比较方法,使用上述相同的还原胺化程序将三种细胞粘附分子包被到市售的醛包被载玻片上。通过目视检查,所有三种表面均能够实现有效的细胞结合(图17b),但是在48h后,仅有DNA缀合的细胞未出现形态变化。可能由于它们表面受体的交联,在这段时间内,ConA和抗CD3固定化的细胞表现出显著的变化。在24和48h后,使用膜联蛋白V和碘化丙啶(PI)染色确定了表面固定的细胞的存活。对于DNA固定的细胞,凋亡和坏死细胞的低百分比与未修饰细胞类似(图17c)。然而,与对照样品相比,ConA和抗CD3固定的细胞显示出明显较大数目的凋亡细胞。因此,DNA分子似乎仅与它们的互补伴侣杂交,而在作用力测量实验中干扰细胞整体生理情况的可能性低得多。
带有所有三种生物分子的AFM尖端容易地捕获活细胞。此捕获简单地通过用悬臂触及细胞膜来完成,其接触时间短至5秒,从而导致细胞向AFM尖端转移。缺少适当生物分子的尖端未捕获细胞。
我们设计了确定悬臂连接强度的测定,得知与基于DNA的细胞和互补官能化的载玻片之间的粘附相比,细胞-悬臂粘附的数目较少,因此整体较弱。由于这种布置,预计细胞-悬臂相互作用将首先被破坏,从而得到了相对较低浓度的生物分子可以实现的相互作用强度。通过实验期间的目视观察,验证了细胞-悬臂相互作用在细胞-表面相互作用之前被破坏。使用两种不同的收缩速率和两种不同的接触力,对每种连接方法测量了去粘附作用力(图18a)。如通过贝尔模型所预测的,对于所有连接方法,所测量的去粘附作用力随接触力和收缩速率而增加。在12%去粘附测量中,ConA连接方法获得了零作用力连接事件。在DNA和抗体情况下,未观察到这类事件。
对于所有三种连接方法,观察到了明显的作用力分布;然而,在所有实验参数下,DNA方法显示出最强的平均粘附,然后是抗体连接,接着是ConA(图18b)。作为对照实验,我们还证明了ConA和抗CD3的捕获效率不受细胞表面上所引入DNA链的存在的影响。应注意对于每种连接策略所确定的整体去粘附作用力取决于连接的数目和收缩速率,并因此不反映单个生物分子相互作用的绝对强度。出于对比,先前已确定分离典型的20bp的DNA双螺旋所需的作用力为38-50pN,这表明如果假设相互作用强度是简单相加的话(尽管多个平行的键可表现出更复杂的换算),那么细胞和悬臂之间大致具有20-25个连接。相似的推理表明涉及了约10个ConA-甘露糖相互作用(每个47pN)和12个抗体-抗原相互作用(每个49pN)。实施了确定每个粘附事件中所涉及的连接数目的实验以更精确地确定这些影响。然而,我们当前的结果表明在典型制备条件下,DNA杂交方法造成了最稳健的连接,尽管每个单独连接的强度可能要小于其他生物分子的强度。
可以通过改变相互作用链的数目和互补区的长度来调节细胞-悬臂相互作用的强度,并且DNA杂交的可逆性还使得所述尖端能够多次使用。这些优势使我们能够使用AFM尖端将细胞每次一个地布置成图案。在最近的报道中,表明个体DNA链可以在印制基底上从一个位置移动到另一个位置,从而允许小分子染料以类似的方式印制。
出于此目的,将5μM的较短DNA链(13个碱基)的溶液包被到悬臂上,并且将80μM的较长链(20个碱基)的溶液偶联到载玻片上。在不依赖CO
概括地说,我们已描述了用于通过AFM研究细胞-细胞相互作用的基于DNA的多用粘附方法的开发。该平台的重要优势包括尖端的重复使用性、相互作用强度的可调节性以及良好限定的化学连接的使用。在所使用的三种基于生物分子的连接策略中,DNA方法在连接后的细胞存活方面表现优异。AFM用于形成个体细胞的精确并且可编程的图案的用途为理解相邻相互作用对细胞分化和调控的影响提供了有用的工具。在先前的报道中,我们已显示可以通过DNA包被的细胞在用互补寡核苷酸印制的表面上的自组装来制备复杂的图案。本发明所述的AFM蘸水笔方法为此技术提供了有用的补充,此技术可以实现产生和调查由多种细胞类型所组成的群集所需的更高分辨率。我们目前正在使用这种方法来阐明在癌症转移、免疫突触形成和细胞-细胞通信中所涉及的基本粘附机理。
用于多价靶向递送载体的病毒衣壳修饰。多价和靶向递送载体为药物施用和诊断成像提供了极大的希望。已经使用了多种核芯支架,其包括聚合物、树枝状聚合物、无机纳米颗粒和脂质体,其在这些应用中已取得了相当大的成功。就基于生物分子的载体而言,还已开发了改造的热休克笼和病毒衣壳,以在其内部容纳药物分子。对于这些载体类型中的每一种,关键性的重要因素是受体结合基团的安装,其能够使载体与靶标组织类型选择性结合。出于此目的,最常用的分子策略包括叶酸、钴胺素、碳水化合物、肽和抗体以及核酸适体。除了将每种分子的多个拷贝连接到不同组成的支架上的期望外,这些分子丰富的化学多样性需要极其耐受官能团的并且在生理条件下进行的化学反应。
已经开发出了数目不断增加的化学选择性偶联反应以用于标记完整尺寸的生物分子。所有报道的方法具有它们特定的优势和理想的用法,并且除了每种技术外,已经出现了用于产生包含多种生物分子成分的复杂结构的新的可能性。为了添加到该列表中,我们报道了在存在高碘酸钠水溶液的情况下,苯胺和苯二胺之间发生的高效氧化偶联反应。到目前为止,该反应显示了出众的化学选择性,并且在微摩尔浓度和在中性pH下快速进行。在该报道中,我们应用这种方法将20-60个DNA适体的拷贝连接到不含基因组的病毒衣壳的表面上。所得的多价组装体与Jurkat细胞表面上的酪氨酸激酶受体结合并且它易于被内吞。最后,我们表明该化学可以与其他生物缀合方法相组合,所述其他生物缀合方法可将功能性药物分子安装在载体内。通过氧化偶联策略制备这些生物杂分子结构的能力很好地预示了它在制备多种不同类型的递送载体中的用途。
噬菌体MS2提供了用于构建靶向递送剂的易于获得的支架。这种病毒的蛋白外壳是由以中空球形结构布置的180个序列相同的单体组成。该外壳蛋白单体可以容易地在大肠杆菌(E.coli)中表达并自组装,从而获得不含基因组的稳健、无毒和生物可降解的结构。参见Carrico,Z.M.;Romanini,D.W.;Mehl,R.A.;Francis,M.B.Chem.Commun.2008,1205-1207,并且其作为参考并入本文。由于MS2衣壳具有与衣壳内部连通的32个孔,因此可以使用正交生物缀合反应在内表面和外表面上实现选择性修饰(Hooker,J.M.;Kovacs,E.W.;Francis,M.B.Journal of the American Chemical Society.2004,126,3718-3719;Kovacs,E.W.;Hooker,J.M.;Romanini,D.W.;Holder,P.G.;Berry,K.E.;Francis,M.B.Bioconjugate Chemistry.2007,18,1140-1147)。在先前的报道中,我们已显示基于酪氨酸的化学可用于安装F-18PET示踪剂(Hooker,J.M.;O'Neil,J.P.;Romanini,D.W.;Taylor,S.E.;Francis,M.Molecular Imaging and Biology.2008,10,182-191)和Gd基MRI造影剂(Hooker,J.M.;Datta,A.;Botta,M.;Raymond,K.N.;Francis,M.B.NanoLetters.2007,7,2207-2210;Datta,A.;Hooker,J.M.;Botta,M.;Francis,M.B.;Aime,S.;Raymond,K.N.Journal of the American Chemical Society.2008,130,2546-2552)以使其用于成像应用。
为了赋予衣壳特异性靶向能力,我们已开发了将核酸适体连接到它们表面上的有效合成方法。使用SELEX(指数富集的配体系统进化技术)方法,可以使适体序列进化以结合几乎任何靶标(Tuerk,C;Gold,L.Science.1990,249,505-510;Ellington,A.D.;Szostak,J.W.Nature.1990,346,818-822),一旦鉴定了组成,它们就可以使用自动化固相合成技术容易地得到。它们的合成还适于引入可改善稳定性或赋予新功能的经修饰主链。另外,适体通常可以达到或甚至超过抗体的特异性和亲和力,并且还外加有尺寸较小的便利性。这些品质使它们成为用于开发具有广泛变化的靶向能力的靶标治疗剂和成像平台的诱人工具。
为了在MS2衣壳的表面上安装寡核苷酸适体,我们选择了先前报道的NaIO
为了开发反应条件,首先选择了20个碱基的DNA序列(链A)。从该序列的胺末端形式开始,通过含NHS-酯前体的酰化作用,容易地引入了N,N-二乙基-N-酰基苯二胺部分。然后,通过改变反应DNA、高碘酸钠的浓度和反应时间来筛选反应条件(图25)。使用相对于MS2-paF19外壳蛋白(20μM,基于单体)的10-20当量(200-400μM)的DNA缀合物和250当量的高碘酸盐(5mM)在室温下反应1h实现了最佳DNA偶联。当在更高浓度的氯化钠下进行反应时,观察到了偶联效力的进一步提高,这可能是由于较高的离子强度对随着更多的DNA连接而产生的负电荷密度积累的屏蔽能力所引起的。对于链A,SDS-PAGE和考马斯染色以及随后进行的光密度测量表明单链DNA修饰了32%的衣壳单体,相当于每个完整衣壳上有55个链。较长的序列显示出稍低的转化,这很可能是由于空间效应以及静电斥力的增加所造成的。反应后,可以使用截留分子量100kDa的尺寸排阻色谱或离心浓缩器将修饰的衣壳与过量的DNA分离。作为技术说明,必须小心地从样品中移除所有甘油、乙二醇或其他邻位二醇以防止它们与高碘酸盐反应。
如图25c,d中所示,通过透射电子显微镜(TEM)和动态光散射(DLS)表明所得的衣壳保持完整。DLS显示一旦链A与衣壳缀合,其水动力直径(hydrodynamic diameter)增加10.5±0.7nm。当引入互补链时,直径又增加了3.9±1.0nm,这表明当与衣壳外部缀合时,DNA仍能够进行沃森-克里克-富兰克林碱基配对。碱基配对能力还提供证据证明DNA链在整个氧化偶联条件下是稳定的。使用变性衣壳单体,通过使用凝胶迁移测定的SDS-PAGE进一步确认了缀合DNA的碱基配对(图25b)。
对于MS2内表面的官能化,选择了标准的半胱氨酸生物缀合。MS2含有已证明在正常马来酰亚胺生物缀合条件下无法接近的两个天然半胱氨酸。因此,表达了双重突变体(MS2-paF19-N87C)以在内表面上引入半胱氨酸。显示了半胱氨酸突变体的反应性,其中在Alexa Fluor 488马来酰亚胺(AF488)存在下对MS2进行荧光标记。此外,MALDI-TOF MS显示了对单一修饰产物的接近定量的转化。缺少半胱氨酸突变的MS2-paF19衣壳显示在相同条件下无染料掺入。
为了完成图24中所示递送载体的合成,选择了靶向Jurkat细胞上特异性细胞表面标志物的41个核苷酸的DNA适体(链B)作为外部靶向基团。使用细胞-SELEX方法分离了先前报道为sgc8c的链B,并且其结合伴侣确定为蛋白质酪氨酸激酶7(PTK7)。PTK7是在Jurkat T白血病细胞以及多种其他白血病细胞系的表面上存在的跨膜蛋白,其已被提议作为T细胞急性淋巴母细胞性白血病的潜在生物标志物。使用氧化偶联策略,将二乙基苯二胺标记的链B的20-40个拷贝连接到每个衣壳上,如通过SDS-PAGE和密度测定分析所确定的(图25a,泳道8)。为了检测细胞结合测定中的衣壳,在DNA连接前,用如上所述的AF488生色团修饰了内部。
通过在37℃下在培养基中与Jurkat细胞一起孵育30-60分钟,我们测试了这些衣壳的靶向特异性。随后使用流式细胞术的分析显示与背景细胞的自荧光相比,暴露于具有链B(在衣壳中11nM)的MS2衣壳的细胞样品表现出平均荧光强度的显著提高,图26a。对于阴性对照,我们合成了无外部修饰的经AF488修饰的衣壳(AF488-MS2)以及用41-nt的随机序列链修饰的衣壳(C)。两种对照衣壳均不能引起平均荧光强度的提高,从而确认了特异性适体序列在细胞结合中的作用。
在流式细胞术实验中验证了B衣壳的靶向性,我们用共聚焦显微镜研究了经修饰衣壳的细胞内化作用。在37℃下与Jurkat细胞一起孵育30-60分钟后,可检测作为细胞内明亮荧光点形式的链B标记的衣壳的存在,图3b。使用荧光内吞标志物共染色的试验表明B标记的衣壳与低密度脂蛋白(LDL)颗粒共定位,而不与转铁蛋白共定位。尽管已知转铁蛋白和LDL均是内吞标志物,但是一旦进入到细胞内部,它们通过不同的途径输送。已显示转铁蛋白指向通过再循环途径靶向回到表面的内体(endosome),而与LDL有关的囊泡最终进入溶酶体。结合α-PTK7适体的靶向特异性,对于溶酶体酸化后优先释放的酸不稳定药物前体的靶向药物递送来说,B-衣壳的溶酶体结局是令人鼓舞的。
对于任何递送载体组合物,未保护的生物分子靶向剂的连接对于实现组织特异性来说将可能是至关重要的。本发明证明了出于此目的的化学选择性氧化偶联反应的应用。原则上,现在可以将本发明所述的基于MS2的载体靶向已确定了结合适体的任何受体。出于诊断成像的目的,对于衣壳来说内化可能不是必需的;然而,设想所观察到的吸收对于药物递送应用是非常有益的。在当前的实验中,我们向这些载体中加入了抗癌药物以及可用于确定体内细胞标志物位置的放射性核素和对比试剂。
一般程序和材料。除非另作说明,否则所有化学品和溶剂均为分析纯并且如从商业来源中所获得时一样地使用。在EM Reagent0.25mm硅胶60-F254板上实施分析薄层色谱(TLC),并通过254nm下的紫外(UV)辐照或用高锰酸钾染色显像。使用旋转蒸发器在减压条件下除去所有有机溶剂。在氮气氛下,从氢化钙中蒸馏二氯甲烷(CH2Cl2)。使用NANOpure纯化系统(Barnstead,USA)对生物过程中使用的或作为反应溶剂使用的水(dd-H2O)去离子。使用先前报道的方法1制备4-(4-二乙基氨基-苯基氨基甲酰基)-丁酸琥珀酰亚胺酯。所有寡核苷酸均得自Integrated DNA Technologies(Coralville,IA)。通过反相HPLC或NAP-5凝胶过滤柱(GE Healthcare)纯化样品。使用LAB CONCO Freezone 4.5(Lab Conco)冻干样品。将冻干的寡核苷酸在适当的缓冲液中再悬浮并通过测量260nm下的吸光值来确定浓度。除非另作说明,否则所有细胞培养试剂均得自Gibco/Invitrogen Corp(Carlsbad,CA)。使用标准技术进行细胞培养。Jurkat细胞在T-25培养瓶(Corning,USA)中在添加了10%(v/v)胎牛血清(FBS,HyClone)和1%青霉素/链霉素(P/S,Sigma)的RPMI培养基1640中生长。
仪器和样品分析NMR。使用Bruker AVQ-400(400MHz)光谱仪测量了1H和13C光谱。将化学位移报告为相对于氯仿-d(δ7.26,s)的δ,以百万分之一(ppm)为单位。如下所述报告多重性:s(单重峰),d(双重峰),t(三重峰,q(四重峰)、dd(双二重峰),p(五重峰),m(多重峰),br(变宽)或app(表观)。将耦合常数报告为以赫兹(Hz)为单位的J值。给定共振的质子数(n)表示为nH,并且基于光谱的积分值。
质谱。在Voyager-DETM系统(PerSeptive Biosystems,USA)上实施了基质辅助激光解吸-电离时间-飞行质谱分析法(MALDI-TOF MS)。在进行MALDI-TOF MS分析前,使用C18
高效液相色谱(HPLC)。在Agilent 1100系列HPLC系统(Agilent Technologies,USA)上实施HPLC。使用在线二极管阵列检测器(DAD)实现了所有HPLC实验的样品分析。使用C18固定相和MeCN/100mM三乙基乙酸铵(TEAA,pH=7.0)梯度完成了寡核苷酸的分析和制备反相HPLC。
凝胶分析。对于蛋白质分析,按照Laemmli的方案,在Bio-Rad(Hercules,CA)的Mini-Protean装置上进行十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)。将所有蛋白电泳样品在1,4-二硫苏糖醇(DTT)存在下在100℃加热10分钟以确保还原任何二硫键。将凝胶在30V运行5分钟,在120V运行70-90分钟以使得条带能够良好分离。将市售标志物(Bio-Rad)加样到每块凝胶的至少一个泳道上以用于指明表观分子量。通过用考马斯亮蓝R-250(Bio-Rad)染色使蛋白质条带显像。对于荧光蛋白缀合物,在UV背光上完成显像。在EpiChem3暗室系统(UVP,USA)上进行凝胶成像。
动态光散射。使用Malvern Instruments Zetasizer Nano ZS,usage courtesyof Jean Frechet获得DLS测量值。根据三次测量的平均值计算数据曲线和标准偏差,每次测量由分别在45秒进行的10次运行组成。测量数据表示为强度图,其用较小尺寸的106倍表示较大尺寸。在10mM pH7.0的磷酸盐缓冲液中取样。
透射电子显微镜(TEM)。在UCBerkeley电子显微镜实验室,使用FEI Tecnai 12透射电子显微镜以100kV的加速电压获得TEM图像。在Cressington 108自动溅射涂布机中通过用氩等离子体(在0.1mbar,以40mA进行30s)使碳包被的聚醋酸甲基乙烯脂支持的铜丝网带电从而制备TEM网格。通过将5μL样品吸取到这些网格上并使它们平衡3分钟从而制备了用于TEM分析的蛋白质样品。然后,用滤纸吸取样品并用ddH2O漂洗。然后,将网格暴露于作为负染色的5μL 1%(w/v)的醋酸双氧铀水溶液90s。除去过度染色后,将网格在空气中干燥。
将苯二胺加入到寡核苷酸中的一般实验程序。购买了在5′-末端含有伯胺的DNA寡核苷酸。典型反应如下所示:将浓度为300μM的DNA与4-(4-二乙基氨基-苯基氨基甲酰基)-丁酸琥珀酰亚胺酯(60-120当量)在DMF和50mM pH8.0的磷酸盐缓冲液(1:1)中的溶液反应。将反应混合物简单地涡旋混合,然后使其在室温下反应2h。按照厂商提供的方案,可以使用RPHPLC或市售凝胶过滤柱从DNA中纯化小分子。纯化后,将DNA冻干,然后在所需的缓冲液中再悬浮。通过测量260nm下的吸光值确定了浓度。A、B和C的序列同一性如下所示:
A:5’-TCATACGACTCACTCTAGGGA-3’
B:5’-ATCTAACTGCTGCGCCGCCGGGAAAATACTGTACGGTTAGA-3’
C:5’-CCCTAGAGTGAGTCGTATGACCCTAGAGTGAGTCGTATGAA-3’
DNA与MS2缀合的一般程序。在eppendorf管中装入MS2-paF19或MS2-paF19-N87C(20μM)、含苯二胺的寡核苷酸(200-400μM)和NaIO
MS2突变体的克隆和表达。先前已报道了pBAD-MS2-paF19质粒的产生和生长。对于p-氨基苯丙氨酸(paF)掺入所必须的tRNA-和tRNA合成酶编码质粒,我们要感谢PeterSchultz实验室(Scripps Research Institute,LaJolla,CA)。使用下列正向和反向引物将87位突变为半胱氨酸:正向:5'-AGC CGC ATG GCG TTC GTA CTT ATG TAT GGA ACT AACCAT TC-3'反向:5'-GAA TGG TTA GTT CCA TAC ATA AGT ACG AAC GCC ATG CGG CT-3’。MS2-paF19-N87C的生长和纯化与MS2-paF19的相同,尽管对于MS2-paF19-N87C所获得的产率较低(约1-10mg/L,相比于MS2-paF19的约20mg/L)。
MS2-paF19-N87C的双表面修饰。首先,在内部半胱氨酸上修饰MS2-paF19-N87C。对于半胱氨酸烷基化反应,将Alexa Fluor 488马来酰亚胺(Invitrogen)(15μL 19mM的DMSO溶液)添加到MS2-paF19-N87C(285μL 100μM在10mM pH7.2磷酸盐缓冲液中的溶液)中。将反应简单地涡旋混合,然后使其在室温下反应1h。通过凝胶过滤(NAP-5)除去过量的小分子并且使用离心过滤浓缩剩余的蛋白质。重要的是要注意在使用前预漂洗离心过滤器,这是因为我们发现这将防止与氧化偶联步骤有关的问题。如上所述,进行外部修饰。
荧光衣壳的流式细胞术。使用标准488Ar激光,在FACSCalibur流式细胞仪(BDBiosciences,USA)上获得流式细胞术分析。对于所有试验,收集了至少10000个活细胞的数据。在培养基中用荧光衣壳(2μM)处理Jurkat细胞(1×10
共聚焦显微镜检查。使用63×Achroplan IR油浸物镜,在Zeiss LSM510 META/NLOAxioimager上进行共聚焦荧光成像。通过在37℃用修饰的MS2(20μM)和DiILDL(15μg/mL,Invitrogen)或Alexa Fluor 594-转铁蛋白(25μg/mL,Invitrogen)孵育细胞30-60分钟对Jurkat细胞进行双重标记。在488nm激发修饰的MS2(用Alexa Fluor488荧光标记),在495-530nm之间收集发射。用543nm的光激发DiI-LDL和AF594-转铁蛋白并且分别在590-625nm和590-655nm之间收集发射。
尽管出于使理解清楚的目的已经通过例证说明和实施例说明了上述发明,但本领域的技术人员将理解可以在所附的权利要求的范围内进行某些变化和改进。另外,本文所提供的每篇参考文献均以每篇参考文献单独作为参考并入本发明的相同程度以其全部作为参考并入。在本发明申请和本发明所提供的参考文献存在矛盾的情况下,应以本发明申请为准。
序列
SEQ ID NO:1
GTA ACC ATC CAG CTG TCA CT
SEQ ID NO:2
ACT GAC ACC TGG ATC GTT AC
SEQ ID NO:3
TCA TAG GAG TCA CTC TAG CG
SEQ ID NO:4
CCC TAG ACT GAG TCG TAT GA
SEQ ID NO:5
ACT GAC TGA CTG ACT GAG TC
SEQ ID NO:6
CAG TCA GTC ACT GAG TCA CT
SEQ ID NO:7
TCATACGACTCACTCTAGGGA
SEQ ID NO:8
ATCTAACTGCTGCGCCGCCGGGAAAATACTGTACGGTTAGA
SEQ ID NO:9
CCCTAGAGTGAGTCGTATGACCCTAGAGTGAGTCGTATGAA
- DNA-细胞缀合物
- 一种DNA-蛋白质缀合物及其在检测汞离子浓度中的应用