小蠹虫信息素的合成方法
文献发布时间:2023-06-19 12:24:27
技术领域
本发明属于化工生产技术领域,特别涉及小蠹虫信息素的合成方法。
背景技术
小蠹虫属鞘翅目小蠹科,我国已知的有500多种,世界范围内,大概有6000多种,其中为害树木的小蠹虫有两种类型:一种类型称为树皮小蠹虫,它们直接蛀食树木韧皮部与边材淀粉纤维,使植物机体成为直接受害者;另一种类型称为蛀干小蠹虫,它们不直接取食植物机体,而是在树木体中构筑坑道,并将真菌孢子带入,使其在坑道周边萌发,好像是有意耕耘,长出来的菌丝和再生孢子,就成了它们的食物。
小蠹虫主要分布在热带、亚热带和温带地区,其在国内淮河以南各省区均有发生;其多发生在枝干或伐倒木、仓储粮仓、器材库,危害稻谷、薯干及竹木器材等。蠹虫遍布我国南北各省区,可危害马尾松、赤松、华山松、油松、樟子松、黑松等,以成虫和幼虫蛀害松树嫩梢、枝干或伐倒木,凡被害梢头,易被风吹折断。
经研究发现化合物(1S,4R)-4-异丙基-1-甲基-2-环己烯-1-醇(CAS:53399-74-9)是小蠹虫的信息素;利用该信息素可以对小蠹虫进行实施诱捕,一则能够监控小蠹虫发生的时间和密度,从而指导如何进行防控;二则能够直接进行捕杀小蠹虫;该方法绿色环保,避免了高毒农药的使用。
有鉴于此,寻找一种理想的小蠹虫信息素(1S,4R)-4-异丙基-1-甲基-2-环己烯-1-醇的合成方法是非常有意义的。
发明内容
为解决上述技术问题,本发明的目的在于提供一种小蠹虫信息素的合成方法,该合成方法所采用的合成原料廉价易得,反应条件温和,操作简单,生产成本低,生产效率高,产率高,产品纯度高,适合放大生产。
为实现上述技术目的,达到上述技术效果,本发明通过以下技术方案实现:
小蠹虫信息素的合成方法,该小蠹虫信息素为(1S,4R)-4-异丙基-1-甲基-2-环己烯-1-醇,该合成方法包括如下步骤:
步骤(1),将环糊精类物质与式(Ⅰ)所示的(S)-(-)-柠檬烯形成包合物,然后在催化剂作用下进行催化氢化反应,得到式(Ⅱ)所示的化合物A;
步骤(2),在含溴盐和氧化剂的溶液体系中,化合物A发生加成反应,得到式(Ⅲ)所示的化合物B;
步骤(3),在大位阻碱作用下,化合物B发生消去反应,得到式(Ⅳ)所示的目标产物(1S,4R)-4-异丙基-1-甲基-2-环己烯-1-醇;
合成路线如下:
优选的,环糊精类物质与(S)-(-)-柠檬烯在溶剂条件下经搅拌形成包合物,溶剂为水、甲醇、乙醇、丙醇、异丙醇、丁醇、异戊醇、环己醇、丙三醇中的至少一种。
优选的,所述环糊精类物质为β-环状糊精、α-环状糊精、γ-环状糊精、羟丙基-β-环状糊精、麦芽糖基-β-环状糊精中的至少一种。
优选的,所述催化剂为钯碳、铂、氧化铂、W1型镍、W2型镍、W3型镍、W4型镍、W5型镍、W6型镍中的至少一种。
优选的,(S)-(-)-柠檬烯、环糊精类物质、催化剂三者的质量比为1:1~1.5:0.005~0.1。
优选的,所述含溴盐为溴化钠、溴化钾、溴化铵、溴化锂、溴化镍、溴化铜、溴化铁、溴化钙、溴化铝、四甲基溴化铵、四乙基溴化铵、四丙基溴化铵、四丁基溴化铵、苄基三甲基溴化铵、苄基三乙基溴化铵、苄基三丁基溴化铵中的至少一种;所述氧化剂为次氯酸钠、氯酸钠、高氯酸钠、溴酸钠、过硫酸氢钾复合盐、双氧水、高锰酸钾、氯酸钾、过硼酸钠、重铬酸钾、高碘酸钠中的至少一种。
进一步的,步骤(2)中,化合物A、含溴盐、氧化剂三者的摩尔比为1:1~1.5:1~1.5。
步骤(3)中,大位阻碱优选为叔丁醇钾、叔丁醇钠、环己醇钠、异戊醇钠、二异丙基氨基锂、双(三甲基硅基)氨基钠、叔丁醇锂、异辛醇钠、异癸醇钠、2-庚醇钠、2-癸醇锂、2-苯乙醇锂中的至少一种。
进一步的,步骤(3)中,化合物B与大位阻碱的摩尔比为1:1~2.5。
进一步的,步骤(1)的反应温度为-20~30℃;步骤(2)的反应温度为20~100℃;步骤(3)的反应温度为-20~50℃。
本发明的有益效果是:
本发明的合成方法在第一步反应中,先利用(S)-(-)-柠檬烯和环糊精形成包合物,再进行催化氢化反应,如此能够抑制(S)-(-)-柠檬烯环内碳碳双键的加氢还原,只使末端碳碳双键进行了加氢还原;在第二步反应中,利用氧化剂氧化溴负离子生成次溴酸,次溴酸对环内碳碳双键进行高选择性加成反应得到溴代叔醇;在第三步反应中,利用大位阻碱为试剂,能够抑制分子内亲核取代反应的发生而抑制生成环氧化合物,只发生溴原子离去的消去反应,从而生成目标产物(1S,4R)-4-异丙基-1-甲基-2-环己烯-1-醇。
该合成方法所采用的合成原料廉价易得,反应条件温和,操作简单方便,生产成本低,生产效率高,产率高,产品纯度高,适合放大生产。
附图说明
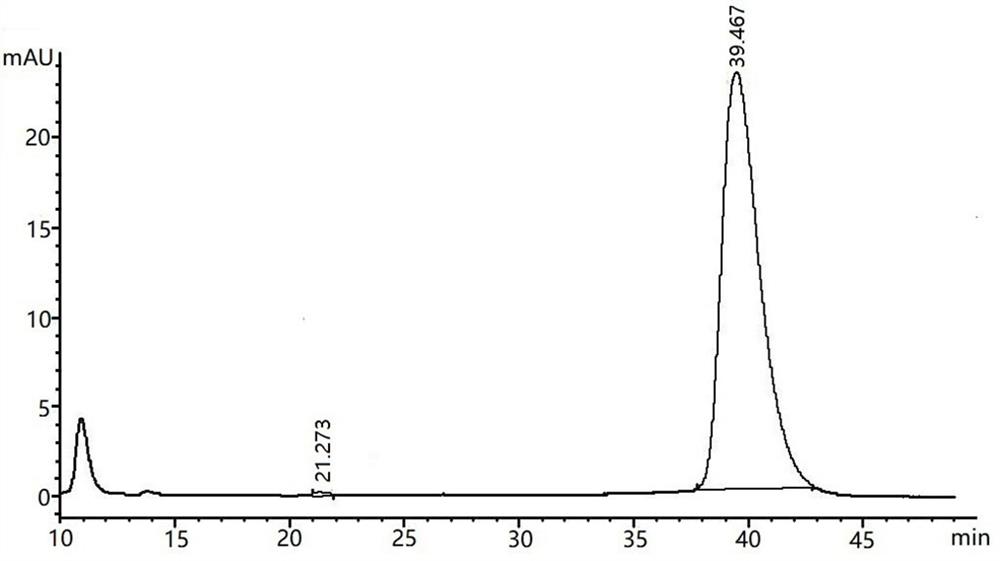
图1为本发明实施例1对应的手性衍生物液相图。
图2为外消旋衍生物液相图。
具体实施方式
下面将结合具体实施例对本发明中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明提供了一种小蠹虫信息素的合成方法,该小蠹虫信息素为(1S,4R)-4-异丙基-1-甲基-2-环己烯-1-醇,具体的,该合成方法包括如下步骤:
步骤(1),将环糊精类物质与式(Ⅰ)所示的(S)-(-)-柠檬烯形成包合物,然后在催化剂作用下进行催化氢化反应,得到式(Ⅱ)所示的化合物A;
步骤(2),在含溴盐和氧化剂的溶液体系中,化合物A发生羟卤加成反应,得到式(Ⅲ)所示的化合物B;
步骤(3),在大位阻碱作用下,化合物B发生消去反应,得到式(Ⅳ)所示的目标产物(1S,4R)-4-异丙基-1-甲基-2-环己烯-1-醇;
合成路线如下:
步骤(1)中,环糊精类物质与(S)-(-)-柠檬烯是在溶剂条件下经搅拌形成包合物,溶剂优选为水、甲醇、乙醇、丙醇、异丙醇、丁醇、异戊醇、环己醇、丙三醇中的至少一种;环糊精类物质优选为β-环状糊精、α-环状糊精、γ-环状糊精、羟丙基-β-环状糊精、麦芽糖基-β-环状糊精中的至少一种;所述催化剂优选为钯碳、铂、氧化铂、W1型镍、W2型镍、W3型镍、W4型镍、W5型镍、W6型镍中的至少一种;(S)-(-)-柠檬烯、环糊精类物质、催化剂三者的质量比为1:1~1.5:0.005~0.1;步骤(1)的反应温度为-20~30℃。
步骤(2)中,所述含溴盐优选为溴化钠、溴化钾、溴化铵、溴化锂、溴化镍、溴化铜、溴化铁、溴化钙、溴化铝、四甲基溴化铵、四乙基溴化铵、四丙基溴化铵、四丁基溴化铵、苄基三甲基溴化铵、苄基三乙基溴化铵、苄基三丁基溴化铵中的至少一种;所述氧化剂优选为次氯酸钠、氯酸钠、高氯酸钠、溴酸钠、过硫酸氢钾复合盐、双氧水、高锰酸钾、氯酸钾、过硼酸钠、重铬酸钾、高碘酸钠中的至少一种;化合物A、含溴盐、氧化剂三者的摩尔比为1:1~1.5:1~1.5;步骤(2)的反应温度为20~100℃。
步骤(3)中,大位阻碱优选为叔丁醇钾、叔丁醇钠、环己醇钠、异戊醇钠、二异丙基氨基锂、双(三甲基硅基)氨基钠、叔丁醇锂、异辛醇钠、异癸醇钠、2-庚醇钠、2-癸醇锂、2-苯乙醇锂中的至少一种;在该步骤中,化合物B与大位阻碱的摩尔比为1:1~2.5;步骤(3)的反应温度为-20~50℃;且该步骤(3)的反应过程是在一定量的有机溶剂中进行;该有机溶剂优选为四氢呋喃、甲基四氢呋喃、N,N-二甲基甲酰胺、异丙醇、丁醇、异戊醇、环己醇、2-庚醇、2-癸醇、甲基环戊基醚、甲基叔丁基醚、六甲基磷酰三胺、N,N-二乙基甲酰胺、乙二醇二甲醚中的至少一种。
实施例1
步骤(1),1#反应瓶中加入50克(S)-(-)-柠檬烯、300毫升乙醇和100毫升蒸馏水,搅拌下加入60克β-环状糊精,冷却至0℃搅拌1小时;然后加入0.3克氧化铂,使用高纯氮气置换3次,然后以每分钟1毫升的速度在液面下通入氢气,使用气相检测跟踪反应进程。
气相检测反应体系中原料含量低于1%时,停止反应,过滤除去固体,滤液浓缩,加入250毫升水,加热至50℃搅拌1小时,使用乙酸乙酯萃取2次;合并有机相,使用50℃水洗涤2次,再经硫酸钠干燥,过滤,浓缩,剩余物使用玻璃真空精馏柱进行常压精馏提纯,收集168-172℃馏分,得到46.6克化合物A,92%收率,99.5%纯度。
步骤(2),2#反应瓶中加入10.3克溴化钠、10克四丁基溴化铵和250毫升水,搅拌溶清,加入13.8克化合物A;控温30℃,滴加15克氯酸钠/50毫升水溶液;滴加完毕,继续在30℃搅拌反应4小时。加入250毫升水稀释反应液,使用乙酸乙酯萃取2次,合并有机相,食盐水洗涤2次,硫酸钠干燥,过滤,浓缩,剩余物经柱层析提纯(层析柱中装有200目硅胶,洗脱剂为石油醚:乙酸乙酯=10:1),得22.3克化合物B,95%收率,99.5%纯度。
步骤(3),氮气保护下,3#反应瓶中加入2.4克氢化钠制成品(60%氢化钠分散于矿物油中,相当于含1.44g纯NaH)和100毫升甲基四氢呋喃,冰水冷却,慢慢注入6.6克异戊醇,待体系无气体生成,冷却至-20℃;在此过程中,NaH与异戊醇生成异戊醇钠;在30分钟内再向3#反应瓶中注入12克化合物B/50毫升甲基四氢呋喃溶液,于-20℃搅拌4小时,然后室温搅拌过夜。
冰水冷却,加入50毫升水萃灭反应,分液,有机相使用硫酸钠干燥,过滤,浓缩,剩余物经柱层析提纯(层析柱中装有400目硅胶,洗脱剂为石油醚:乙酸乙酯=80:1),得6.8克小蠹虫信息素化合物,88%收率,99.6%纯度,光学纯度99.4%;对产物进行检测分析:
实施例2
步骤(1),1#反应瓶中加入50克(S)-(-)-柠檬烯、500毫升乙醇,搅拌下加入50克羟丙基-β-环状糊精,冷却至5℃搅拌1小时;然后加入5克W6型镍,使用高纯氮气置换3次,然后以每分钟1毫升的速度在液面下通入氢气,使用气相检测跟踪反应进程。
气相检测反应体系中原料含量低于1%时,停止反应,过滤除去固体,滤液浓缩,加入250毫升水,加热至50℃搅拌1小时,使用乙酸乙酯在50℃萃取2次;合并有机相,50℃水洗涤2次,再经硫酸钠干燥,过滤,浓缩,剩余物使用玻璃真空精馏柱进行常压精馏提纯,收集168-172℃馏分,得到45.6克化合物A,90%收率,99.1%纯度。
步骤(2),2#反应瓶中加入14.7克溴化铵和500毫升水,搅拌溶清,加入13.8克化合物A;控温30℃,剧烈搅拌下在1小时内分批加入92克 过硫酸氢钾复合盐(每次1克左右);加料完毕,继续在30℃下搅拌反应2小时。冷却至0℃,抽滤,滤液使用乙酸乙酯萃取2次,合并有机相,食盐水洗涤2次,硫酸钠干燥,过滤,浓缩,剩余物经柱层析提纯(层析柱中装有200目硅胶,石油醚:乙酸乙酯=10:1),得23克化合物B,98%收率,99.6%纯度。
步骤(3),氮气保护下,3#反应瓶中加入2.4克氢化钠制成品(60%氢化钠分散于矿物油中,相当于含1.44g纯NaH),冰水冷却,慢慢注入100克2-庚醇,待体系无气体生成,冷却至0℃;在此过程中,NaH与2-庚醇生成2-庚醇钠;在30分钟内再向3#反应瓶中注入12克化合物B/50毫升2-庚醇溶液,于0℃搅拌反应过夜。
冰水冷却,加入200毫升水萃灭反应,使用正己烷萃取5次,合并有机相,饱和食盐水洗涤2次,硫酸钠干燥,过滤,浓缩,剩余物经柱层析提纯(层析柱中装有400目硅胶,洗脱剂为石油醚:乙酸乙酯=80:1)得6.9克小蠹虫信息素化合物,90%收率,99.8%纯度,光学纯度99.8%。
实施例3
该实施例3与实施例2的区别在于:步骤(3)不同;实施例3的步骤(3)如下:
氮气保护下,3#反应瓶中加入100克2-癸醇,冰水冷却,慢慢注入25毫升正丁基锂溶液(2.5M,溶剂为正己烷),待体系无气体生成,冷却至-20℃;在此过程中,正丁基锂与2-癸醇反应生成2-癸醇锂;然后在30分钟内再注入12克化合物B/50毫升2-癸醇溶液,于-20℃搅拌反应过夜。
冰水冷却,加入500毫升水萃灭反应,使用正己烷萃取5次,合并有机相,饱和食盐水洗涤2次,硫酸钠干燥,过滤,浓缩,剩余物经柱层析提纯(层析柱中装有400目硅胶,洗脱剂为石油醚:乙酸乙酯=80:1)得6.8克小蠹虫信息素化合物,89%收率,99.5%纯度,光学纯度99.7%。
上述实施例中的目标产物(1S,4R)-4-异丙基-1-甲基-2-环己烯-1-醇的光学纯度检测方法如下:
首先制备衍生物,向微量反应瓶中加入20毫克所得的(1S,4R)-4-异丙基-1-甲基-2-环己烯-1-醇、50毫克碳酸铯和2毫升DMF(N,N-二甲基甲酰胺),搅拌10分钟,加入30毫克2,5-二氟硝基苯,室温搅拌过夜;把反应液倒入50毫升水中,乙醚萃取3次,合并有机相,饱和食盐水洗涤2次,硫酸钠干燥,过滤,浓缩,剩余物经柱层析提纯(层析柱中装有200目硅胶,洗脱剂为石油醚:乙酸乙酯=10:1),得手性衍生物;对该手性衍生物利用手性液相色谱法进行光学纯度检测;手性液相色谱系统采用Chiralpak AS手性柱,20%异丙醇/正己烷流动相。衍生物的获得路线如下:
取20mg外消旋化合物((1S,4R)-4-异丙基-1-甲基-2-环己烯-1-醇和(1R,4R)-4-异丙基-1-甲基-2-环己烯-1-醇1:1的混合物),利用上述同样的衍生物制备方法,获得外消旋衍生物,并利用手性液相色谱法进行检测。
如图1所示为实施例1对应的手性衍生物液相图,图2为外消旋衍生物液相图;对液相图进行分析,可得手性衍生物的光学纯度。
以上所述仅为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书及附图内容所作的修改或等效变换,或直接或间接运用在其他相关的技术领域,均同理包括在本发明的专利保护范围内。
- 小蠹虫信息素的合成方法
- 以受控方式分配小蠹虫信息素的装置