一种聚草酸丁二醇酯的原位制备方法
文献发布时间:2023-06-19 18:32:25
技术领域
本发明属于高分子材料合成技术领域,具体的说,涉及一种能同时提高聚草酸丁二醇酯结晶温度、结晶速率和特性粘度,并降低其色度的原位制备方法。
背景技术
PAOX是一类草酸基聚酯材料的统称,通常是以草酸二甲酯/草酸二乙酯单体为原料,和不同二元醇通过缩合聚合反应制备得到,常见的用来参与聚合反应的醇类单体如乙二醇、1,3-丙二醇、1,4-丁二醇等脂肪族二元醇和1,4-环己烷二甲醇、异山梨醇等环状二元醇。目前在众多的草酸基脂肪族聚酯材料中,聚草酸乙二醇酯(PEOX)、聚草酸丁二醇酯(PBOX)由于其高熔点以及良好的降解性能而获得了广泛的关注。
与聚草酸乙二醇酯(PEOX)相比,虽然聚草酸丁二醇酯(PBOX)熔点稍低,为100℃左右,但是作为为数不多的熔点能达到100℃以上的脂肪族聚酯材料,PBOX也有其独特的性能,如良好的延展性、较高的断裂伸长率和断裂强度以及耐热性和降解性能,可以在一定程度上缓解市面上可降解材料空缺的局面。
专利CN102219891B涉及一种聚草酸-1,4-丁二醇酯的制备方法,具体为:将草酸二乙酯和丁二醇在控制氮气流速的情况下在80-170℃下恒温搅拌0.5-5h后得到酯化产物,其中醇酯比为1-3,催化剂优选为草酸,后进一步的在高温110-180℃下、真空200-500Pa下恒温恒压反应3-8h可得到目标产物。其特性粘度仅为0.05-0.45dL/g,熔点和结晶温度仅为104.8℃和55.9℃,并且产物都是暗白色固体。
专利US.Pat.4,186,189A中,公开了以草酸二乙酯和1,4-丁二醇为原料,先在常压下进行酯交换反应,后在减压条件下进行缩聚反应。物质的量比为1:1.15的草酸二乙酯和1,4-丁二醇,加入1%的钛酸四(2-乙基己醇)酯为催化剂,在氮气保护下分别在140℃和160℃恒温反应2h,减压阶段在2-3mm汞柱压强,160℃和180℃分别恒温恒压反应20h和2h。产物的特性粘度为0.95d L/g,Tm为105℃。此方法反应条件苛刻,成本非常高,反应时间长达26h,不适合工业生产。
在聚酯的合成过程中,绝大部分商品化聚酯是通过二元羧酸和二元醇的熔融缩合聚合方式合成的,具有低成本、单体来源丰富且可完全转化、无需使用有机溶剂等优点。二元羧酸和二元醇的熔融缩聚基于可逆的酸醇酯化反应,其分子量控制同时受到热力学和动力学的影响。在热力学上,通过高温和高真空等手段将酯化反应产生的副产物水或者甲醇等小分子从体系中排出,从而可将其转化成不可逆反应。制约聚酯分子量的另一个因素是其动力学特点,即要求精确控制官能团等摩尔比进行反应。而在实际操作过程中很难实现精确的基团等摩尔比,即使反应起始时单体纯度达到很高,称量准确,在反应过程中也可能会因为单体蒸发或升华速率不同和各种副反应导致的羧基或者羟基的消耗而偏离等摩尔比。因此,传统的酯化方法获得的聚酯产物分子量不高,要实现高分子量聚酯的合成,就必须实现反应过程中官能团的动态等摩尔比。基本都是单体按化学计量配置,加微量单官能团物质或使某双官能团原料单体过量来控制分子量,但是这种方法一方面会造成原料的浪费,增加了生产成本,另一方面由于过量的单体会加大副反应的程度导致对产物性能造成不良的影响。
虽然目前已有研究者关注到了聚草酸丁二醇酯这一领域,但是全部都只停留在实验室小实验容器试验阶段,并未有研究者将其成功放大,也即现有方法距离产业化生产还存在一定差距。而工业放大的实现对聚酯合成配方、合成路线以及实验操作和步骤有着密切的关系,就聚草酸丁二醇酯而言,当以草酸为原料时,草酸的低温就易分解性使得反应难以进行;当以草酸二酯类为原料时,草酸二酯的易升华性会使反应过程中醇酯比发生改变,无法把控好醇酯比的动态平衡。而且在聚草酸丁二醇酯聚合过程中,原料丁二醇极易发生丁二醇醚化生成四氢呋喃的副反应,并且在放大过程中丁二醇的大量增加会使得这种副反应程度加剧,严重影响聚合反应的进行。在聚合反应后期,由体系粘度急剧上升而带来的巨大的放热效应会使得体系温度不断升高,进而使副反应程度进一步加深并且产物在高温下也会容易热分解。所以从实验室小瓶实验阶段到逐步放大的过程极其艰难。
针对以上现有技术中聚草酸丁二醇酯反应时间按长、特性粘度低并且颜色差的缺陷,开发一种能得到低色度和高特性粘度的同时,又能提高结晶速率的聚草酸丁二醇酯的制备方法尤为迫切,不仅提高了聚草酸丁二醇酯的使用范围,并且在一定程度上能缓解可降解材料短缺的困境。
发明内容
针对以上现有技术中制备聚草酸丁二醇酯存在的反应时间按长、特性粘度低、结晶温度低且颜色差的缺陷,开发一种聚草酸丁二醇酯的原位制备方法,得到的聚草酸丁二醇酯具有低色度和高特性粘度的性能,其结晶温度和熔点也显著提高,不仅提高了聚草酸丁二醇酯的使用范围,在一定程度上能缓解可降解材料短缺的困境。
第一方面,本发明提供一种聚草酸丁二醇酯的原位制备方法,以草酸二甲酯/草酸二乙酯和1,4-丁二醇为原料,在钛-磺酸复合催化剂和第二类金属氧化物的作用下,依次经过酯交换反应、预缩聚和终缩聚阶段,即得到聚草酸丁二醇酯,制得的聚草酸丁二醇酯呈亮白色,具有结晶度高、特性粘度高的特点;所述钛-磺酸复合催化剂在酯交换反应前加入;所述第二类金属氧化物在酯交换反应后、预缩聚反应前加入。具体的反应方程式如下所示:
其中n为草酸丁二醇酯结构单元的数目,n>100,n为大于100的整数,例如100、101、102、103、104、105、106、……200、……300等。
在反应前,投料时严格控制草酸二甲酯/草酸二乙酯与1,4-丁二醇的摩尔比为1比1,保证投料时体系内酯基与羟基基团数比
所述钛-磺酸复合催化剂的加入量为草酸二甲酯/草酸二乙酯质量分数的0.5-1‰;所述第二类金属氧化物用量为草酸二甲酯/草酸二乙酯质量分数的1-3‰。
进一步地,作为关键技术环节:
在酯交换反应前,将反应原料草酸二甲酯或草酸二乙酯、1,4-丁二醇和钛-磺酸复合催化剂一起加入到反应釜中,以10℃/min的升温速率将体系升温至70-80℃下搅拌打浆60min以上;优选为80℃。
所述的酯交换温度为120℃-160℃,优选为160℃,以出第一滴小分子甲醇/乙醇为计时零点,酯交换反应时间为180-300min。
所述的预缩聚温度为120℃-130℃,优选为130℃,预缩聚反应时间为30min-60min,优选为45min,预缩聚压力为绝对压强为2kPa-3kPa。
所述的终缩聚温度为185℃-195℃,优选为185℃,终缩聚时间为120min-240min,优选为180min,终缩聚压力为绝对压强为100Pa以下。
进一步地,所述的第二类金属氧化物为二氧化钛、三氧化二锑、醋酸锑中的至少一种。
进一步地,所述的钛-磺酸复合催化剂采用以下方法制备,具体步骤包括:
S1:在低温(0-5℃)下,将含钛元素化合物和磺酸类化合物加入到无水的稀释液里,机械搅拌30min以上;
S2:将体系恢复至室温(20-30℃),继续搅拌20min以上;
S3:升高温度至稀释液沸点以上20℃,蒸出多余的稀释液和反应产生的小分子后,自然降温至室温(20-30℃),得到浅黄色液体,即得到所述钛-磺酸复合催化剂。
进一步地,所述的含钛元素化合物选自钛酸四丁酯、钛酸四异丙酯、钛酸四叔丁酯中的至少一种;所述磺酸类化合物选自甲基磺酸、对甲苯磺酸、苯磺酸、三氟甲磺酸、乙基磺酸、对羟基苯磺酸、间羟基苯磺酸、邻甲苯磺酸、氨基磺酸中的至少一种。
进一步地,所述含钛元素化合物和磺酸类化合物的摩尔比为0.5-2,优选为1。
进一步地,所述的稀释液选自无水甲醇、无水乙醇的至少一种,优选为无水乙醇。
进一步地,所述的稀释液与含钛元素化合物、磺酸类化合物质量和的比为1-2,优选为1.5。
进一步地,所述步骤S1中机械搅拌30-60min,搅拌转速为80r/min。
进一步地,所述步骤S2中搅拌时间20-30min。
以钛酸四丁酯和甲基磺酸为原料制备出的钛-磺酸复合催化剂,化学反应式如下:
如上所述的聚草酸丁二醇酯材料的特性粘度为1.4-1.8;重均分子量为130-180kDa;L值在80以上;结晶温度为61-73℃;熔点为105-110℃;结晶度为30-60%。
有益效果:
(1)本发明提出的聚草酸丁二醇酯的原位制备方法,所用的钛-磺酸复合催化剂解决了普通钛系催化剂容易水解失活的问题,减少了合成过程中催化剂的用量,提高了经济效益;
(2)本发明的提出的原位制备方法在预缩聚反应阶段加入第二类金属氧化物催化剂,不仅起到催化效果,并且还能为聚草酸丁二醇酯的结晶提供成核位点,起到了原位催化成核的效果;
(3)本发明的一种聚草酸丁二醇酯的原位制备方法,能在较低温度、较短时间内合成高质量的聚草酸丁二醇酯,生产耗能降低。
附图说明
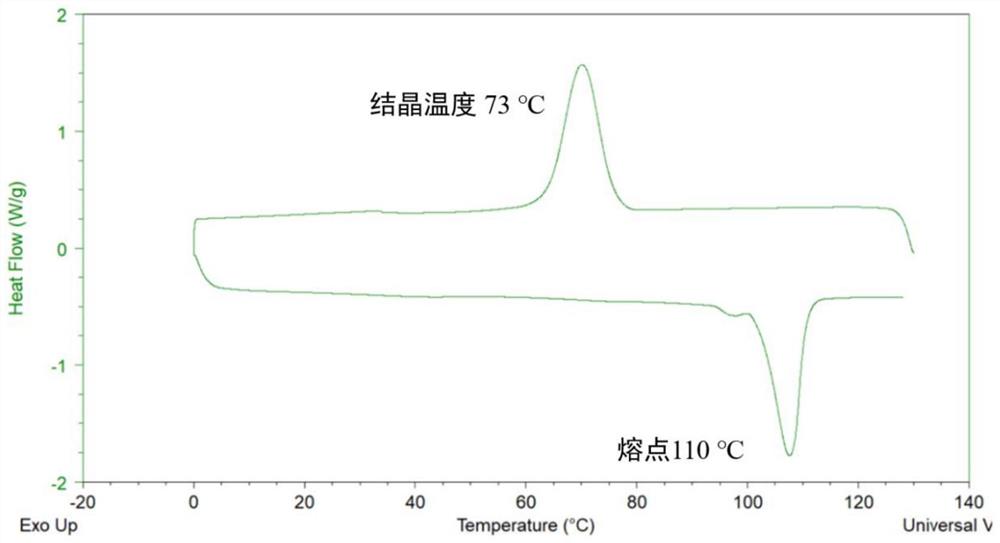
图1为使用原位催化成核剂合成的聚草酸丁二醇酯的DSC图;
图2为普通合成的聚草酸丁二醇酯的DSC图;
图3为使用原位成核剂合成的聚草酸丁二醇酯的核磁共振氢谱图;
图4为使用5L聚合反应釜、原位催化成核剂合成的聚草酸丁二醇酯切粒后图。
具体实施方式
下面结合附图和实施例对本发明进一步说明。
本发明中,特性粘度的测试方法为:按GB/T 14190-2008中5.1.1规定进行。溶剂选用苯酚/1,1,2,2-四氯乙烷(质量比50:50),称样量0.1000g~0.1050g,溶剂10mL。
色度L值的测试方法为:试验按GB/T 14190-2008中5.5.2规定进行。采用CIE1976L*a*b*色系。
结晶温度和结晶热焓采用仪器型号为美国TA公司的DSC25进行DSC测试,并根据测试结果计算聚合物结晶温度和熔点。测试时,采用的程序为:在氮气氛围下(流速为50ml/min),升降温速率都为10℃/min。
催化成核剂微观形貌采用美国FEI公司生产的JSM-7900型场发射扫描电镜对扁桃酸镁进行微观形貌的测试,工作电压与电流分别为230V和8A,测试前需要对样品进行喷金以及吹扫处理。
聚合物的分子量和分子量分布由美国Waters公司带有1515HPLC型泵的Waters-2414凝胶渗透色谱仪表征,流动相为四氢呋喃(THF),测试温度为40℃,以将10mg样品溶解在5ml THF溶剂中,完全溶解后注射过滤器过滤,流量为1.0ml/min,并采用聚苯乙烯(PS)为标样来计算得到分子量。
聚草酸丁二醇酯材料结晶度利用RigakuD/Max-Ultima+型X射线衍射仪测定。
实施例1(钛-磺酸复合催化剂的制备)
(1)在0℃下,将0.1mol(34g)钛酸四丁酯和0.1mol(9.6g)甲基苯磺酸加入到65.4g无水乙醇中,机械搅拌45min,搅拌转速为80r/min;
(2)将体系恢复至室温25℃,继续搅拌30min;
(3)升高温度至无水乙醇沸点以上20℃,蒸出多余的无水乙醇和反应产生的小分子丁二醇后,自然降温至室温25℃,得到浅黄色液体即为耐水解钛-磺酸复合催化剂。
对比实施例1(钛-磺酸复合催化剂中磺酸超出限定范围)
(1)在0℃下,将0.1mol(34g)钛酸四丁酯和0.3mol(28.6g)甲基苯磺酸加入到65.4g无水乙醇中,机械搅拌45min,搅拌转速为80r/min;
(2)将体系恢复至室温25℃,继续搅拌30min;
(3)升高温度至无水乙醇沸点以上20℃,蒸出多余的无水乙醇和反应产生的小分子丁二醇后,自然降温至室温25℃,得到浅黄色液体即为耐水解钛-磺酸复合催化剂。
对比实施例2(钛-磺酸复合催化剂中钛酸物质超出限定范围)
(1)在0℃下,将0.3mol(102g)钛酸四丁酯和0.1mol(9.6g)甲基苯磺酸加入到65.4g无水乙醇中,机械搅拌45min,搅拌转速为80r/min;
(2)将体系恢复至室温25℃,继续搅拌30min;
(3)升高温度至无水乙醇沸点以上20℃,蒸出多余的无水乙醇和反应产生的小分子丁二醇后,自然降温至室温25℃,得到浅黄色液体即为耐水解钛-磺酸复合催化剂。
实施例2(钛-磺酸复合催化剂的制备)
(1)在5℃下,将0.15mol(51g)钛酸四丁酯和0.1mol(9.6g)甲基磺酸加入到66.1g无水乙醇中,机械搅拌30min,搅拌转速为80r/min;
(2)将体系恢复至室温30℃,继续搅拌30min;
(3)升高温度至无水乙醇沸点以上20℃,蒸出多余的无水乙醇和反应产生的小分子丁二醇后,自然降温至室温30℃,得到浅黄色液体即为耐水解钛-磺酸复合催化剂。
实施例3(钛-磺酸复合催化剂的制备)
(1)在0℃下,将0.1mol(28.4g)钛酸四异丙酯和0.1mol(17.2g)对甲苯磺酸加入到68.4g无水乙醇中,机械搅拌45min,搅拌转速为80r/min;
(2)将体系恢复至室温25℃,继续搅拌30min;
(3)升高温度至无水乙醇沸点以上20℃,蒸出多余的无水乙醇和反应产生的小分子丁二醇后,自然降温至室温25℃,得到浅黄色液体即为耐水解钛-磺酸复合催化剂。
实施例4(钛-磺酸复合催化剂的制备)
(1)在2℃下,将0.12mol(40.8g)钛酸四丁酯和0.08mol(13.8g)对甲苯磺酸加入到81.9g无水甲醇中,机械搅拌35min,搅拌转速为80r/min;
(2)将体系恢复至室温25℃,继续搅拌30min;
(3)升高温度至无水甲醇沸点以上20℃,蒸出多余的无水乙醇和反应产生的小分子丁二醇后,自然降温至室温25℃,得到浅黄色液体即为耐水解钛-磺酸复合催化剂。
实施例5(钛-磺酸复合催化剂的制备)
(1)在4℃下,将0.1mol(28.4g)钛酸四异丙酯和0.1mol(15.8g)苯磺酸加入到57.5g无水乙醇中,机械搅拌35min,搅拌转速为80r/min;
(2)将体系恢复至室温25℃,继续搅拌30min;
(3)升高温度至无水乙醇沸点以上20℃,蒸出多余的无水乙醇和反应产生的小分子丁二醇后,自然降温至室温25℃,得到浅黄色液体即为耐水解钛-磺酸复合催化剂。
实施例6(聚草酸丁二醇酯的制备)
(1)将17mol(2007g)草酸二甲酯,17mol(1532g)1,4-丁二醇和1g(0.5‰wt)实施例1中所制备的钛-磺酸复合催化剂加入到5L钢制反应釜中,以10℃/min的升温速率将体系升温至80℃下搅拌打浆60min;
(2)打浆结束后将体系升温至160℃,开始酯交换反应,以出第一滴小分子甲醇/乙醇为计时零点,反应时间为240min;
(3)酯交换结束后,将体系降温至130℃,加入6g(3‰wt)第二类原位催化成核剂三氧化二锑后开始预缩聚反应,体系绝对压强为2kPa,预缩聚时间为45min;
(4)预缩聚结束后,将体系压强调至绝对压强100Pa以下,反应温度升温至185℃,开始终缩聚反应,时间为180min。反应结束后将反应釜内物料熔体在氮气作用下挤出,经过水冷和切粒机造粒后即可得到目标聚酯材料。
将得到的聚草酸丁二醇酯聚酯材料进行测试,得到其重均分子量为180kDa;特性粘度为1.8dL/g;结晶温度为73℃;熔点为110℃;色度L值为87;结晶度为60%。
对比实施例3(无原位催化成核剂)
(1)将17mol(2007g)草酸二甲酯,17mol(1532g)1,4-丁二醇和1g(0.5‰wt)实施例1中所制备的钛-磺酸复合催化剂加入到5L钢制反应釜中,以10℃/min的升温速率将体系升温至80℃下搅拌打浆60min;
(2)打浆结束后将体系升温至160℃,开始酯交换反应,以出第一滴小分子甲醇/乙醇为计时零点,反应时间为240min;
(3)酯交换结束后,将体系降温至130℃后开始预缩聚反应,体系绝对压强为2kPa,预缩聚时间为45min;
(4)预缩聚结束后,将体系压强调至绝对压强100Pa以下,反应温度升温至185℃,开始终缩聚反应,时间为180min。反应结束后将反应釜内物料熔体在氮气作用下挤出,经过水冷和切粒机造粒后即可得到目标聚酯材料。
将得到的聚草酸丁二醇酯聚酯材料进行测试,得到其重均分子量为164kDa;特性粘度为1.65dL/g;结晶温度为54℃;熔点为105℃;色度L值为81;结晶度为42%。
对比实施例4(钛-磺酸催化剂中磺酸过量+无原位催化成核剂)
(1)将2007g(17摩尔)草酸二甲酯,1532g(17摩尔)1,4-丁二醇和1g(0.5‰wt)对比实施例1中所制备的钛-磺酸复合催化剂加入到5L钢制反应釜中,以10℃/min的升温速率将体系升温至80℃下搅拌打浆60min;
(2)打浆结束后将体系升温至160℃,开始酯交换反应,以出第一滴小分子甲醇/乙醇为计时零点,反应时间为240min;
(3)酯交换结束后,将体系降温至130℃后开始预缩聚反应,体系绝对压强为2kPa,预缩聚时间为45min;
(4)预缩聚结束后,将体系压强调至绝对压强100Pa以下,反应温度升温至185℃,开始终缩聚反应,时间为180min。反应结束后将反应釜内物料熔体在氮气作用下挤出,经过水冷和切粒机造粒后即可得到目标聚酯材料。
将得到的聚草酸丁二醇酯聚酯材料进行测试,得到其重均分子量为96kDa;特性粘度为0.87dL/g;结晶温度为47℃;熔点为103℃;色度L值为68;结晶度为36%。
对比实施例5(钛-磺酸催化剂中钛酸物质过量+无原位催化成核剂)
(1)将17mol(2007g)草酸二甲酯,17mol(1532g)1,4-丁二醇和1g(0.5‰wt)对比实施例2中所制备的钛-磺酸复合催化剂加入到5L钢制反应釜中,以10℃/min的升温速率将体系升温至80℃下搅拌打浆60min;
(2)打浆结束后将体系升温至160℃,开始酯交换反应,以出第一滴小分子甲醇/乙醇为计时零点,反应时间为240min;
(3)酯交换结束后,将体系降温至130℃后开始预缩聚反应,体系绝对压强为2kPa,预缩聚时间为45min;
(4)预缩聚结束后,将体系压强调至绝对压强100Pa以下,反应温度升温至185℃,开始终缩聚反应,时间为180min。反应结束后将反应釜内物料熔体在氮气作用下挤出,经过水冷和切粒机造粒后即可得到目标聚酯材料。
将得到的聚草酸丁二醇酯聚酯材料进行测试,得到其重均分子量为86kDa;特性粘度为0.84dL/g;结晶温度为46℃;熔点为103℃;色度L值为67;结晶度为36%。
对比实施例6(单独钛酸四丁酯催化剂+无原位催化成核剂)
(1)将17mol(2007g)草酸二甲酯,17mol(1532g)1,4-丁二醇和1g(0.5‰wt)钛酸四丁酯催化剂加入到5L钢制反应釜中,以10℃/min的升温速率将体系升温至80℃下搅拌打浆60min;
(2)打浆结束后将体系升温至160℃,开始酯交换反应,以出第一滴小分子甲醇/乙醇为计时零点,反应时间为240min;
(3)酯交换结束后,将体系降温至130℃后开始预缩聚反应,体系绝对压强为2kPa,预缩聚时间为45min;
(4)预缩聚结束后,将体系压强调至绝对压强100Pa以下,反应温度升温至185℃,开始终缩聚反应,时间为180min。反应结束后将反应釜内物料熔体在氮气作用下挤出,经过水冷和切粒机造粒后即可得到目标聚酯材料。
将得到的聚草酸丁二醇酯聚酯材料进行测试,得到其重均分子量为136kDa;特性粘度为1.25dL/g;结晶温度为53℃;熔点为103℃;色度L值为73;结晶度为38%。
对比实施例7(单独甲基苯磺酸催化剂+无原位催化成核剂)
(1)将17mol(2007g)草酸二甲酯,17mol(1532g)1,4-丁二醇和1g(0.5‰wt)甲基苯磺酸催化剂加入到5L钢制反应釜中,以10℃/min的升温速率将体系升温至80℃下搅拌打浆60min;
(2)打浆结束后将体系升温至160℃,开始酯交换反应,以出第一滴小分子甲醇/乙醇为计时零点,反应时间为240min;
(3)酯交换结束后,将体系降温至130℃后开始预缩聚反应,体系绝对压强为2kPa,预缩聚时间为45min;
(4)预缩聚结束后,将体系压强调至绝对压强100Pa以下,反应温度升温至185℃,开始终缩聚反应,时间为180min。反应结束后将反应釜内物料熔体在氮气作用下挤出,经过水冷和切粒机造粒后即可得到目标聚酯材料。
将得到的聚草酸丁二醇酯聚酯材料进行测试,得到其重均分子量为141kDa;特性粘度为1.33dL/g;结晶温度为53℃;熔点为104℃;色度L值为74;结晶度为38%。
实施例7(聚草酸丁二醇酯的制备)
(1)将2007g(17摩尔)草酸二甲酯,1532g(17摩尔)1,4-丁二醇和1g(0.5‰wt)实施例3中所制备的钛-磺酸复合催化剂加入到5L钢制反应釜中,以10℃/min的升温速率将体系升温至80℃下搅拌打浆60min;
(2)打浆结束后将体系升温至160℃,开始酯交换反应,以出第一滴小分子甲醇/乙醇为计时零点,反应时间为240min;
(3)酯交换结束后,将体系降温至130℃,加入6g(3‰wt)第二类原位催化成核剂三氧化二锑后开始预缩聚反应,体系绝对压强为2kPa,预缩聚时间为45min;
(4)预缩聚结束后,将体系压强调至绝对压强100Pa以下,反应温度升温至185℃,开始终缩聚反应,时间为180min。反应结束后将反应釜内物料熔体在氮气作用下挤出,经过水冷和切粒机造粒后即可得到目标聚酯材料。
将得到的聚草酸丁二醇酯聚酯材料进行测试,得到其重均分子量为177kDa;特性粘度为1.74dL/g;结晶温度为70℃;熔点为109℃;色度L值为86;结晶度为58%。
实施例8(聚草酸丁二醇酯的制备)
(1)将17mol(2007g)草酸二甲酯,17mol(1532g)1,4-丁二醇和1g(0.5‰wt)实施例1中所制备的钛-磺酸复合催化剂加入到5L钢制反应釜中,以10℃/min的升温速率将体系升温至80℃下搅拌打浆60min;
(2)打浆结束后将体系升温至160℃,开始酯交换反应,以出第一滴小分子甲醇/乙醇为计时零点,反应时间为240min;
(3)酯交换结束后,将体系降温至130℃,加入6g(3‰wt)第二类原位催化成核剂二氧化钛后开始预缩聚反应,体系绝对压强为2kPa,预缩聚时间为45min;
(4)预缩聚结束后,将体系压强调至绝对压强100Pa以下,反应温度升温至185℃,开始终缩聚反应,时间为180min。反应结束后将反应釜内物料熔体在氮气作用下挤出,经过水冷和切粒机造粒后即可得到目标聚酯材料。
将得到的聚草酸丁二醇酯聚酯材料进行测试,得到其重均分子量为165kDa;特性粘度为1.65dL/g;结晶温度为55℃;熔点为105℃;色度L值为83;结晶度为43%。
上述虽然结合附图对本发明的具体实施方式进行了描述,但并非对本发明保护范围的限制,所属领域技术人员应该明白,在本发明的技术方案的基础上,本领域技术人员不需要付出创造性劳动即可做出的各种修改或变形仍在本发明的保护范围以内。
- 一种生物降解性聚(丁二酸乙二醇-co-草酸乙二醇)酯及其制备方法
- 一种聚呋喃二甲酸丁二醇酯和聚对苯二甲酸丁二醇酯共混物的制备方法
- 一种聚呋喃二甲酸丁二醇酯和聚对苯二甲酸丁二醇酯共混物的制备方法