用于制备C2至C3烃的方法
文献发布时间:2023-06-19 18:32:25
相关申请的交叉引用
本申请要求于2020年6月30日提交的美国临时专利申请第63/045,888号的优先权,该美国临时专利申请的全部公开内容特此通过引用并入。背景技术
技术领域
本说明书大体上涉及将含有氢和碳的进料流转化为C
通常,在杂化催化剂方法中,含有氢和碳的进料流诸如合成气体(合成气)包含氢(H
背景技术
对于许多工业应用来说,烃被用于或作为起始材料,来生产塑料、燃料和各种下游化学品。C
用于将进料碳转化为所需产物,如低级烃的合成方法是已知的。然而,当前杂化催化剂方法通常需要合成微孔催化剂组分,其产生可能非常昂贵。
因此,需要可以保持组合C
发明内容
本公开的实施方案通过使用杂化催化剂利用合成气制备C
另外的特征和优点将在以下具体实施方式中进行阐述,并且部分将从该实施方式中对本领域技术人员变得显而易知或通过实践本文所描述的实施方案(包括以下具体实施方式和权利要求书)而认识到。
应当理解,前述整体描述和以下详细描述两者都描述了各种实施方案,并且旨在提供用于理解所要求保护的主题的性质和特征的概述或框架。
附图说明
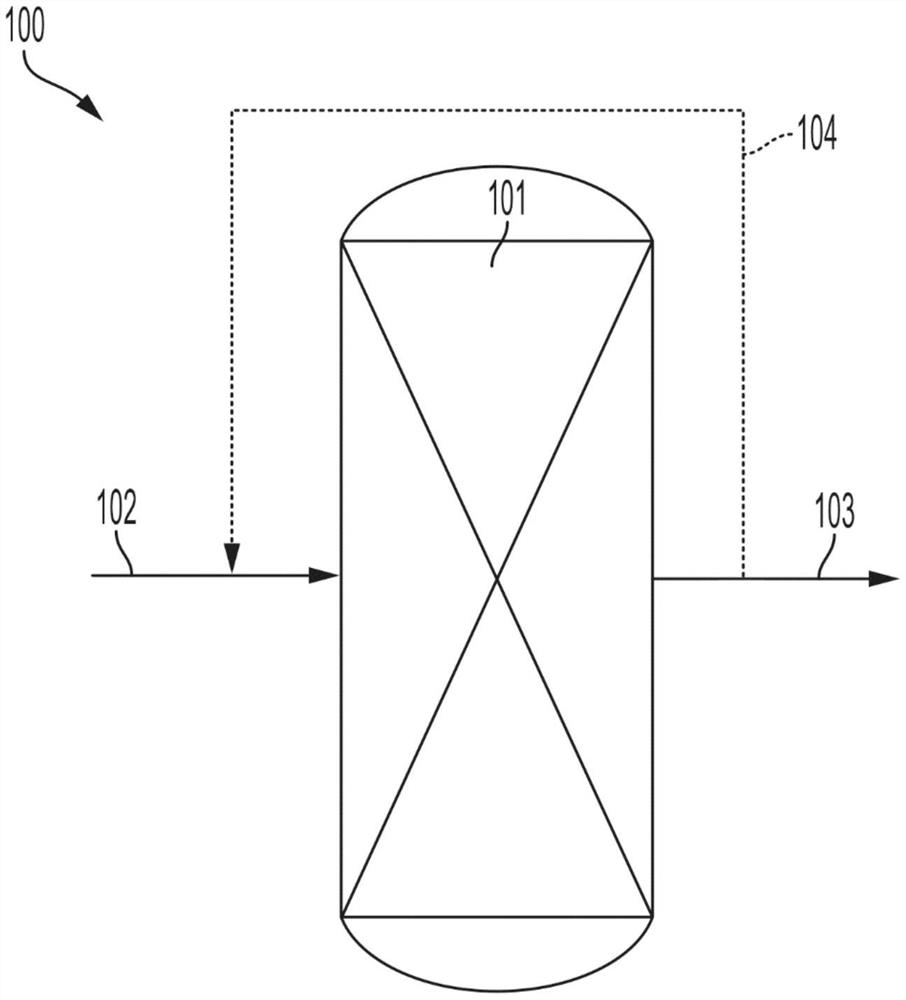
图1是示出根据本公开的一个或多个实施方案的被引入反应器的两个流和离开反应器的一个所得产物流的示意图。
具体实施方式
现在将详细参考利用合成气制备C
如本文所用,需注意“合成气体”和“合成气”在本文中用于表示主要包含氢气、一氧化碳、二氧化碳和非常通常一些惰性物的混合物。
已知当将包含碳的进料流转化为所需产物,例如C
根据本文公开和描述的实施方案的方法解决了产物中的组合C
参考图1的实施方案,进料流102被进料到反应区101中,该进料流102可包含H
如上文所公开,进料流102可包含H
不受任何特定理论束缚,据信焦化和去焦速率几乎平衡,这可能显著减少对杂化催化剂的焦化。这进而可以允许杂化催化剂保留在反应区101中持续延长的时间段而不需要再生。在实施方案中,杂催化剂可保留在反应区101中而无焦化持续大于5小时,诸如大于7.5小时、大于10小时、大于12.5小时、大于15小时、大于17.5小时、大于20小时、大于22.5小时、大于25小时、大于27.5小时或大于30小时。
如本文先前所述,C
现在将描述反应区101内的反应条件。进料流102可以在足以形成包含C
在实施方案中,反应条件还包括反应区101内部的至少20巴(20,000千帕(kPa)),诸如至少25巴(25,000kPa)、至少30巴(30,000kPa)、至少35巴(35.00kPa)、至少40巴(40,000kPa)、至少45巴(45,000kPa)、至少50巴(50,000kPa)、至少55巴(55,000kPa)、至少60巴(60,000kPa)、至少65巴(65,000kPa),或至少70巴(70,000kPa)的压力。在实施方案中,反应条件包括反应区101内部的20巴(20,000kPa)至70巴(70,000kPa),诸如25巴(25,000kPa)至65巴(65,000kPa)、或30巴(30,000kPa)至60巴(60,000kPa)、35巴(35,000kPa)至55巴(55,000kPa)、40巴(40,000kPa)至50巴(50,000kPa)的压力。
在实施方案中,反应条件还包括在反应区101内气时空速(GHSV)(测量为进料流102的体积/催化剂的体积/小时)为至少500h
现在将描述在以上公开的方法中使用的杂化催化剂。如先前所描述,杂化催化剂系统包含将进料流转化为氧化烃的金属氧化物催化剂组分以及将氧化烃转化为烃的微孔催化剂组分。
在一个或多个实施方案中,混合金属氧化物催化剂组分可为本体催化剂或载体催化剂,并且可通过任何合适的方法(如共沉淀、浸渍等)来制备。在实施方案中,混合金属氧化物催化剂组分可包含镓、镧或它们的组合。在实施方案中,混合金属氧化物催化剂组分包含氧化锆。在实施方案中,混合金属氧化物催化剂组分可包含负载在氧化锆上的镓、镧或它们的组合。根据由微孔催化剂组分确定的产物构成,考虑另外的混合金属氧化物催化剂组分。应理解,混合金属氧化物组分混合物中的任何金属均可以多种氧化态存在。还应理解,特定氧化物(例如,Ga
在实施方案中,可通过将混合金属氧化物催化剂组分暴露于常规还原气体而在暴露于进料流102之前在反应器内还原混合金属氧化物催化剂组分。在一个或多个实施方案中,当暴露于进料流102中的还原气体(诸如H
根据实施方案,杂化催化剂包含与微孔催化剂组分混合的混合金属氧化物催化剂组分,该微孔催化剂组分可以选自具有8-MR孔口的分子筛,其中微孔催化剂组分来源于天然矿物。天然矿物可包括菱沸石、伊利石或插晶菱沸石。菱沸石是沸石族的网状硅酸盐矿物,并且可包括菱沸石-Ca、菱沸石-K、菱沸石-Na和菱沸石-Sr。毛沸石是沸石族的纤维矿物。插晶菱沸石是沸石族的水合硅酸盐矿物,并且可包括插晶菱沸石-Na、插晶菱沸石-Ca或插晶菱沸石-Cs。具有8-MR孔口的其它天然存在的矿物也可以用作微孔催化剂组分的来源,包括但不限于斜发沸石或片沸石(骨架类型HEU两者)、钙十字沸石(骨架类型PHI)、辉沸石(骨架类型STI)和钠沸石(骨架类型NAT)。这些矿物可根据2016年矿业年鉴(美国内政部(U.S.Department of the Interior),2018年8月)和IHS化学品经济手册报告“沸石”(12月16日,16 2016)以商业规模生产。
来源于天然矿物的微孔催化剂组分可以优于合成微孔催化剂组分。与合成微孔催化剂组分相比,天然矿物可以是便宜的且易于获得的原料。可被加工为微孔催化剂组分的天然矿物的成本和可用性导致来源于天然矿物的微孔催化剂组分是合成微孔催化剂组分的有吸引力的替代物。另外,如实施例进一步证明,已发现天然矿物可以取代合成微孔催化剂组分并且根据需要表现。
可以加工天然矿物以产生微孔催化剂组分。例如,天然矿物可以与铵盐的水溶液进行离子交换。在另一个实施方案中,可以蒸煮微孔催化剂组分。此外,天然矿物不需要使用有机结构导向剂(SDA)进行合成,如合成微孔催化剂组分所需要。
在实施方案中,微孔催化剂组分可选自CHA、ERI、LEV骨架类型和它们的组合,骨架类型对应于国际沸石协会的命名惯例。在实施方案中,微孔催化剂组分可包含二氧化硅-铝酸盐或硅铝磷酸盐中的一种或多种。应当理解,在实施方案中,可使用硅铝酸盐、硅铝酸盐和硅铝磷酸盐骨架两者。在实施方案中,微孔催化剂组分可以是具有毛沸石(ERI)骨架类型的H-ERI。
根据实施方案,金属组分可包含0.1重量%至10.0重量%的混合金属氧化物催化剂组分。例如,金属组分可以包含0.1重量%至9.0重量%的混合金属氧化物催化剂组分,例如0.1重量%至1.0重量%、0.1重量%至2.0重量%、0.1重量%至3.0重量%、0.1重量%至4.0重量%、0.1重量%至5.0重量%、0.1重量%至6.0重量%、0.1重量%至7.0重量%、0.1重量%至8.0重量%、0.5重量%至1.0重量%、0.5重量%至2.0重量%、0.5重量%至3.0重量%、0.5重量%至4.0重量%、0.5重量%至5.0重量%、0.5重量%至6.0重量%、0.5重量%至7.0重量%、0.5重量%至8.0重量%、0.5重量%至9.0重量%、0.5重量%至10.0重量%、1.0重量%至2.0重量%、1.0重量%至3.0重量%、1.0重量%至4.0重量%、1.0重量%至5.0重量%、1.0重量%至6.0重量%、1.0重量%至7.0重量%、1.0重量%至8.0重量%、1.0重量%至9.0重量%、或1.0重量%至10.0重量%。在实施方案中,金属组分可包含2.0重量%至10.0重量%的混合金属氧化物催化剂组分,诸如2.0重量%至3.0重量%、2.0重量%至4.0重量%、2.0重量%至5.0重量%、2.0重量%至6.0重量%、2.0重量%至7.0重量%、2.0重量%至8.0重量%、2.0重量%至9.0重量%、3.0重量%至4.0重量%、3.0重量%至5.0重量%、3.0重量%至6.0重量%、3.0重量%至7.0重量%、3.0重量%至8.0重量%、3.0重量%至9.0重量%、或3.0重量%至10.0重量%。在一个或多个实施方案中,金属组分可包含4.0重量%至10.0重量%的混合金属氧化物催化剂组分,诸如4.0重量%至5.0重量%、4.0重量%至6.0重量%、4.0重量%至7.0重量%、4.0重量%至8.0重量%、4.0重量%至9.0重量%、5.0重量%至6.0重量%、5.0重量%至7.0重量%、5.0重量%至8.0重量%、5.0重量%至9.0重量%、或5.0重量%至10.0重量%。
根据实施方案,微孔催化剂组分可包含小于或等于50.0的SiO
这些的示例可以包括但不必限于:选自ERI实施方案和LEV实施方案。还可以采用具有以上骨架类型中的任一种的微孔催化剂组分的组合。应当理解,取决于期望产物,微孔催化剂组分可以具有不同元环孔开口。例如,取决于期望产物,可以使用具有8-MR至12-MR孔开口的微孔催化剂组分。然而,为了产生C
不受任何特定理论束缚,据信孔口尺寸(Atlas of zeolite framework types,第6版,Elsevier,第381-86页,2007)和笼限定环尺寸(ACS Catalysis,2019,第9卷,第6017页)可以有助于所需烃产物。孔口尺寸对于缩小产物分布可能是重要的。另外,笼限定环尺寸对于增强产物分布中的C
如以下实施例进一步证明,微孔催化剂组分在与混合金属氧化物催化剂组分组合之前可以进行离子交换。不受任何特定理论束缚,离子交换可以通过经由用氢置换金属原子(诸如但不限于钾或钠)增加微孔催化剂组分的酸度来改变微孔催化剂组分(并且因此,整体杂化催化剂)的特性。另外,微孔催化剂组分可以通过本领域技术人员已知的方法蒸煮或去铝。由于微孔催化剂组分的酸度降低,因此这可以改变杂化催化剂的化学性质,以调整所述方法以形成期望的产物。
杂化催化剂的混合金属氧化物催化剂组分和微孔催化剂组分可以通过任何合适的方式混合在一起,例如通过物理混合—如摇动、搅拌或其他搅动。在实施方案中,混合金属氧化物催化剂组分和微孔催化剂组分可作为单一配制的催化剂存在。混合金属氧化物催化剂组分和微孔催化剂组分可以0.1:1至10:1(如0.5:1至9:1)范围内的重量/重量(wt/wt)比(混合金属氧化物催化剂组分:微孔催化剂组分)存在于反应区101中,典型地作为杂化催化剂存在于催化剂床中。
虽然本文所述的杂化催化剂可以适用于除合成气体到烃型系统以外的方法,但是已发现,杂化催化剂可不在各种方法之间直接转移。也就是说,催化剂可以是一种方法的有效催化剂,但在另一种方法中可能显示性能较差。例如,在甲醇至烯烃的方法中显示具有令人满意的性能的催化剂可能无法直接转移到合成气体到烃的方法中,其中同一种催化剂可能不表现出相同的令人满意的性能。此外,关于其它方法例如甲醇至烯烃(MTO)方法中的产物分布的公开和教导可能不可转移到合成气体到烃型系统中。例如,包含具有CHA拓扑的SSZ-13的杂化催化剂在不同系统(诸如MTO)中可能未展示提供合成气体到烃型系统的相同产物分布。
通过以下实施例进一步阐明实施方案。
在实施例1中,微孔催化剂组分由毛沸石制备。从内华达州伊斯特盖特(Eastgate,Nevada)收集来源于Minerals Research,P.O.Box 591,Clarkson,NY 14430的毛沸石。将大致10g的毛沸石25220放置在具有搅拌棒的燧石玻璃罐中。然后,添加100mL 0.1M乙酸铵。执行一系列三次离子交换。在每次离子交换后,通过过滤回收固体,冲洗,然后再悬浮。在第一次交换中,在环境温度下将毛沸石和乙酸铵搅拌十八个小时。在第二次交换中(过滤、冲洗和再悬浮后),在环境温度下将毛沸石和乙酸铵搅拌五小时。在第三次交换中(过滤、冲洗和再悬浮后),再次在环境温度下将毛沸石和乙酸铵搅拌十八小时。在第三次且最后一次交换后,通过过滤收集最终产物,将其冲洗,然后在90℃下干燥。干燥后,通过将温度以2.5℃/分钟升至550℃来在空气中煅烧产物。然后将温度在550℃下保持四小时。然后将煅烧的粉末压实并将尺寸设定成60-100目径,以形成H-ERI微孔催化剂组分。
为了制备混合金属氧化物催化剂组分,将4.40mL镓储备溶液(去离子水中具有C=2.0M的硝酸镓(III)水合物)、1.76mL的镧储备溶液(去离子水中具有C=1.5M的六水合硝酸镧(III))和2.86mL去离子水的浸渍溶液混合。然后,将20g具有100m
为了制备杂化催化剂,将100mg的H-ERI微孔催化剂组分与50mg的混合金属氧化物催化剂组分混合并且振荡三十秒。
在实施例2中,使用与实施例1相同的程序制备微孔催化剂组分和混合金属氧化物催化剂组分。当制备杂化催化剂时,将112.5mg的H-ERI微孔催化剂组分与37.5mg的混合金属氧化物催化剂组分混合并且振荡三十秒。
为了制备微孔催化剂组分,进一步处理实施例1的H-ERI微孔催化剂组分以产生LZ-220微孔催化剂组分。LZ-220进一步描述于美国专利第4,503,023号中,该专利以引用方式并入本文。首先,通过将8.0g(NH
以与实施例1相同的方式制备混合金属氧化物催化剂组分。最后,将100mg的LZ-220微孔催化剂组分与50mg的混合金属氧化物催化剂组分混合并且振荡三十秒。
在实施例4中,使用与实施例3相同的程序制备微孔催化剂组分和混合金属氧化物催化剂组分。当制备杂化催化剂时,将112.5mg的LZ-220微孔催化剂组分与37.5mg的混合金属氧化物催化剂组分混合并且振荡三十秒。
在实施例5中,为了制备微孔催化剂组分,将3g天然菱沸石置于500mL烧杯中。然后,将300mL去离子水中的1M NH
为了制备混合金属氧化物催化剂组分,将4.0mL镓储备溶液(去离子水中具有C=2.0M的硝酸镓(III)水合物)、3.852mL的镧储备溶液(去离子水中具有C=0.623M的六水合硝酸镧(III))和0.148mL去离子水的浸渍溶液混合。然后,将15g具有100m
然后通过将145.9mg的H-CHA微孔催化剂组分与216.8mg的混合金属氧化物催化剂组分组合来制备杂化催化剂并且振荡三十秒。
在实施例6中,使用与实施例5相同的程序制备微孔催化剂组分和混合金属氧化物催化剂组分。当制备杂化催化剂时,将150.8mg的H-CHA微孔催化剂组分与98.1mg的混合金属氧化物催化剂组分混合并且振荡三十秒。
在实施例7中,使用与实施例5相同的程序制备微孔催化剂组分。
为了制备混合金属氧化物催化剂组分,将2.87mL的铟储备溶液(去离子水中具有C=1.553M的硝酸铟(III)水合物)、2.118mL的镧储备溶液(去离子水中具有C=0.623M的六水合硝酸镧(III))和3.011mL去离子水的浸渍溶液混合。然后,将15g具有100m
然后通过将151.4mg的H-CHA微孔催化剂组分与100.7mg的混合金属氧化物催化剂组分组合来制备杂化催化剂并且振荡三十秒。
在实施例8中,使用与实施例5相同的程序制备微孔催化剂组分。
混合金属氧化物催化剂组分是可商购的Cu基甲醇合成催化剂HiFuel
然后通过将150.4mg的H-CHA微孔催化剂组分与102.1mg的混合金属氧化物催化剂组分组合来制备杂化催化剂并且振荡三十秒。
在实施例9中,使用与实施例5相同的程序制备微孔催化剂组分。
为了制备混合金属氧化物催化剂组分,将0.983mL锌储备溶液(去离子水中具有C=2.5M的硝酸锌(II)水合物)和1.867mL去离子水的浸渍溶液混合。然后,将1g具有140m
然后通过将155.2mg的H-CHA微孔催化剂组分与100.2mg的混合金属氧化物催化剂组分组合来制备杂化催化剂并且振荡三十秒。
在实施例10中,使用与实施例5相同的程序制备微孔催化剂组分。
为了制备混合金属氧化物催化剂组分,将4.754mL锌储备溶液(去离子水中具有C=1.473M的硝酸锌(II)水合物)和3.246mL去离子水的浸渍溶液混合。然后,将10g具有100m
然后通过将147.7mg的H-CHA微孔催化剂组分与101.9mg的混合金属氧化物催化剂组分组合来制备杂化催化剂并且振荡三十秒。
在实施例11中,使用与实施例5相同的程序制备微孔催化剂组分。
混合金属氧化物催化剂组分是可商购获得的具有70m
然后通过将151.1mg的H-CHA微孔催化剂组分与101.2mg的混合金属氧化物催化剂组分组合来制备杂化催化剂并且振荡三十秒。
为了制备SAPO-34微孔催化剂组分,根据美国专利4,440,871A的程序合成微孔催化剂组分,该专利以引用方式并入本文。然后通过将温度以5℃/分钟从25℃升温到600℃来在空气上煅烧所形成的微孔催化剂组分。将温度在600℃下保持四小时,然后在四小时的时间段内将温度降至25℃。然后将煅烧的粉末压实并将尺寸设定成60-100目径,以形成SAPO-34微孔催化剂组分。
在比较例1中,混合金属氧化物催化剂组分以及杂化催化剂的制备与实施例1相同。通过将50mg微孔催化剂组分与100mg混合金属氧化物催化剂组分组合并摇动30秒来形成杂化催化剂。
在比较例2中,使用比较例2的相同微孔催化剂组分和相同混合金属氧化物催化剂组分形成杂化催化剂。通过将112.5mg微孔催化剂组分与37.5mg混合金属氧化物催化剂组分组合并摇动30秒来形成杂化催化剂。
在比较例3中,微孔催化剂组分是SSZ-13。按照Chemical Communications 2016,42,3227中描述的合成来制备微孔催化剂组分。更具体地,将41.2g高纯度水添加到具有搅拌棒的塑料烧杯中。然后将13.6g的25重量%的模板氢氧化物溶液添加到塑料烧杯中,随后添加0.96g的50重量%NaOH溶液。搅拌该混合物。然后,添加0.32g Al(OH)
比较例4的微孔催化剂组分是被粒化并设定尺寸为60-80目径的天然矿物菱沸石。
在比较例4中,使用与实施例5相同的混合金属氧化物催化剂组分。通过将117.1mg微孔催化剂组分与214.9mg混合金属氧化物催化剂组分组合并摇动30秒来形成杂化催化剂。
在比较例5中,微孔催化剂组分与实施例5中形成的微孔催化剂组分相同。对于混合金属氧化物催化剂组分,使用可商购获得的具有140m
在比较例5中,通过将151.1mg微孔催化剂组分与101.2mg混合金属氧化物催化剂组分组合并摇动30秒来形成杂化催化剂。
表1中示出了实施例1-11和比较例1-5的微孔催化剂组分的组成。
在内径为3mm的管状不锈钢反应器或内径为2mm的石英固定床反应器中进行催化测试。不锈钢反应器的底部具有用于保持催化剂床的金属玻璃料。石英反应器底部装有顶部有羊毛的石英片,以固定催化剂床。在催化测试中,将杂化催化剂装载到催化剂上并且遵循以下程序:随着温度从25℃升高到反应温度(以5℃/分钟增加),氮气流动到反应器。类似地,压力从环境条件增加到反应压力。然后,用合成气体流替换氮气流。将合成气体流继续冲洗一小时,之后开始气相色谱分析。在设定的运行时间持续时间之后,当反应器从反应温度和压力返回到环境温度和压力时,合成气流被氮气流替换。
在实施例1-11和比较例1-5的催化测试中利用两种不同的反应条件,其示出于表2中。
最后,表3中示出了实施例1-11和比较例1-6的各种催化测试的催化数据。另外,表4中进一步详述了产物的选择性。使用气相色谱分析产物。定期进行组分(N
从表3和4中可以看出,包含8-MR孔开口小于或等于
一氧化碳转化率(X
其中F
将i组分的选择率[C mol%]测量为表3和4中指定的运行时间的所有数据点的平均值。用于计算i组分的选择率的公式示出于以下等式2中:
其中n
碳平衡率(CB)[C mol%]是以一氧化碳形式进入反应区的碳总量与以一氧化碳和含碳产物形式离开反应区的碳之间的比率。用于计算碳平衡率的公式示出于等式3中:
其中n
C
其中F
对于本领域技术人员将显而易见的是,在不脱离要求保护的主题的精神和范围的情况下,可对本文所述的实施方案作出各种修改和变化。因此,本说明书旨在覆盖本文所述的各个实施方案的修改和变化,条件是这些修改和变化落入所附权利要求书和其等效物的范围内。
- 用于催化木质素模型化合物反应制备芳香烃的催化剂及制备方法
- 用于生产C2和C3烃的方法
- 用于产生C2和C3烃的方法