一种由精液介导的转基因鸡输卵管生物反应器的制备方法
文献发布时间:2023-06-19 18:32:25
技术领域
本发明涉及基因工程技术领域,具体是一种由精液介导的转基因鸡输卵管生物反应器的制备方法。
背景技术
转基因动物生物反应器的出现克服了细菌基因工程产物的缺陷和细胞基因工程生产规模的限制,从而受到人们极大的关注。家禽具有周期短、生产性能高、研究成本低,而且表达产物更接近天然状态和易于纯化等优点。1993年英国罗斯林研究所Sang研究禽类蛋黄表达系统,在禽蛋卵黄成功表达外源蛋白,但含量不高。1994年曾邦哲(曾杰)最早提出了禽类转基因输卵管生物反应器(oviduct bioreactor)的概念(以及术语和词汇),并在1996年北京召开的第1届国际转基因动物学术研讨会上阐述了系统(结构)遗传学方法与转基因生物技术研究。在此期间,中国曾邦哲与加拿大、美国、英国、日本有关转基因禽类实验室和禽类开发的企业联系,但国际上当时没有人开展这项课题。美国Avigenics公司和Georgia大学R.Ivarie教授与曾邦哲(曾杰)展开了通信探讨研究后独自开展了立项和规模化投资与开发。目前,国际上有十多家前景看好的公司以转基因输卵管生物反应器作为拳头开发产品,约2003年英国罗斯林研究所也创建了公司,并由Sang主持研究课题,从禽类蛋黄表达系统转向了输卵管生物反应器。转基因输卵管生物反应器,采用蛋清蛋白质基因的侧翼特异表达调控序列、启动子和多肽分泌信号序列,构建在禽类输卵管细胞特异性表达外源贵重蛋白质的转基因生物反应器,蛋清蛋白质在输卵管细胞表达、含量比较高、蛋白质修饰质量好。输卵管生物反应器(Oviduct Bioreactor)成为继哺乳动物乳腺生物反应器后最具发展前景的动物生物反应器。
生产转基因鸡的方法可以通过显微注射,将外源基因直接注射到单细胞受精卵细胞质中,经过体外培养后,产生转基因鸡。但是,此种方法取到一个受精卵需要一只母鸡,若操作失败,一个细胞的损失就相当于一只母鸡的损失,成本较高,并且受精卵体外孵化条件也很复杂,操作难度较大。在国内,李赞东等将脂质体包装后的质粒pMiwz(含有报告基因LacZ)注入囊胚期胚盘中,转基因获得成功,并证明囊胚期注射外源基因可以获得转染的原生殖细胞(PGCs),并能够传给下一代。Jamie love等将带有目的基因质粒DNA注射入鸡受精卵的胚胎中,体外培养12h后发现近一半存活,40%胚胎中含有质粒DNA,有6%相当于每一个细胞含有1个拷贝的外源基因,7只存活至性成熟雏鸡中的一只将3.4%外源基因传递给其后代。此种方法可以得到转基因嵌合体鸡,但转基因细胞的嵌合部位和嵌合程度不容易确定。逆转录病毒载体利用重组的逆转录病毒载体DNA包装成高滴度的病毒颗粒,将此病毒颗粒注射到囊胚腔内或将去除透明带的胚胎与分泌病毒的细胞共培养,使携带外源基因的反转录病毒RNA在感染过程中整合到宿主细胞的染色体内,达到外源基因转移到胚胎的目的病毒正常生命周期特征来感染家禽,从而导入外源并达到整合的目的。使用的病毒载体有两类:复制完全型和复制缺陷型(高波,2000)。复制完全型逆转录病毒包含全部结构基因能自我复制,并能重复感染细胞。而复制缺陷型逆转录病毒缺乏部分或全部结构基因,不能自我复制,需在辅助细胞中繁殖。反转录载体法的整合效率比电穿孔和脂质体转染法要高,但是由于基因片段大小的限制和获得高滴度病毒有困难,所以主要停留在实验室阶段,还未广泛应用于商业生产(刘向萍,2003)。
与显微注射法和体细胞核移植等转基因方法相比,精子载体法具有简便易行、耗费低、易于规模制备等一系列优点,在转基因家畜上显示了诱人的应用前景。精子介导法介导生产转基因动物可分为体外导入和体内导入两种方法,体外导入是直接采用外源DNA或用阳离子脂质体包埋外源DNA与成熟精子共孵育后,通过人工授精获得转基因禽类;体内导入是睾丸注射法,是将外源DNA直接注射到雄性动物的睾丸内,使得精子携带外源DNA,然后通过动物自然交配或者人工授精使的整合了外源DNA的精子与卵子结合从而得到含有外源DNA的转基因动物。睾丸注射法相较于体外直接孵育的优势是能够避免体外操作对精子的活力造成影响,但是禽类不同于哺乳动物,禽类睾丸在腹腔内,睾丸注射法还需要对雄性动物做开刀手术,对动物会有一定的伤害,对于雄性禽类,虽然附睾中不含有抑制外源DNA进。入精子是精清,但是在实际睾丸注射的过程中直接将外源DNA注射入附睾还是很困难的,对操作技术要求较高,同样不利于技术的推广。这种方法利用精卵结合的正常生理过程来完成外源基因的导入,具有简便易行,对卵原核无损伤等特点。但各种精子与外源DNA共孵育后转染的方法可重复性不高且对受精能力有一定影响,整合能力也较差,且其具体机制尚不清楚。
基因编辑是指可以对特定的基因组定点修饰的新技术,包括基因定点插入、敲除、突变,目前主导的基因编辑技术包括锌指蛋白核酸酶(Zinc-fingemuclease,ZFNs)、转录激活因子样效应核酸酶(Transcriptionactivator-likeeffectornucleases,TALFENs)和通过sgRNA引导的CRIAPR/Cas9核酸酶。相较于ZFNs和TALENs,CPISPR/Cas9核酸酶成本低、制作简单、实验周期短、作用更加高效、靶点分布广且可以同时对多个基因进行编辑,所以得到了更加广泛的应用。CRISPR系统由三部分组成:CRISPR主体基因座(CRISPR locus),基因座上游的一段前导序列(leader sequence,LS)及CRISPR功能相关的基因(CRISPR-associated genes,Casgenes)[54]。CRISPR序列上游的序列启动CRISPR转录出未成熟的crRNA,与tracrRNA结合并成熟,进而与Cas蛋白形成蛋白核酸复合体系,可以引导Cas蛋白靶向目标序列实现特异性识别、结合与双链切割。目前使用最广泛的是由II型CRISPR系统改造而来CRISPR/Cas9系统,通过人工设计sgRNA,引导Cas9蛋白对特异序列识别并切割,造成DNA双链断裂(double strand break,DSB)。DSB就会促使细胞启动DNA损伤修复机制:非同源末端连接(non-homologous end joining,NHEJ)和同源重组修复(homology-directedrepair,HDR),从而实现基因序列的插入、缺失、替换。对于SgRNA,RNA聚合酶III识别的U6或H1启动子在启动转录时依赖的聚合酶在RNA的5'端需要G或GG,所以在设计sgRNA的5'端无G或GG时,如何在合成有活性的sgRNA的同时,又提高Cas9核酸酶编辑的效率是目前研究者需要克服的一大难题。
发明内容
本发明的目的在于克服现有技术的不足,提供一种由精液介导的转基因鸡输卵管生物反应器的制备方法,以至少达到提高转基因效率以及筛选具有高水平表达的个体的目的。
本发明的目的是通过以下技术方案来实现的:一种由精液介导的转基因鸡输卵管生物反应器的制备方法,包括以下步骤:
S1、设计靶向鸡基因组DNA的gRNA:选用鸡基因组目标DNA序列为卵清蛋白,选择同源性90%以上的gRNA;
S2、构建gRNA的表达载体;
S3、构建携带有外源基因表达框的供体载体,将供体载体转变为ssDNA;
S4、将载体通过精子载体法人工授精导入鸡个体。
本发明通过将CRISPR基因编辑技术和精子载体法结合,在两种技术的协同作用下,不仅提高了外源基因整合到基因组中的准确率,还实现了高效率导入外源基因,制备出能够表达并分泌外源蛋白到鸡蛋蛋清中的转基因鸡,并实现外源基因稳定的遗传给后代。
进一步的,步骤S2包括以下步骤:
根据PAM规则确定靶位点序列后,合成符合表达载体构建要求的靶位点序列正负DNA单链,退火形成DNA双链之后连接到CRISPR/Cas系统表达载体上,载体导入细胞后能表达gRNA和Cas9蛋白。
进一步的,步骤S2还可以通过以下方法实现,构建到已有的gRNA表达载体上:单独构建gRNA表达质粒,启动子采用U6,T7等Polll聚合酶,与Cas9表达质粒,或者Cas9蛋白共转染到目标细胞。
进一步的,PAM类型包括5'-NNN-3'、5'-NNNN-3'、5'-NNNNN-3'、5'-NNNNNN-3'、5'-NNNNNNN-3'和5'-NNNNNNNN-3'。
进一步的,步骤S3包括以下步骤:
以以步骤S2中形成的双链DNA作为同源重组修复模板,将步骤S1中的目标DNA位点DSB处上游和下游各100bp-1000bp序列,分别连接在外源基因的5'端和3'端,作为同源臂,将供体载体转变为ssDNA。
进一步的,步骤S3中所述外源基因表达框包括一段补全卵清蛋白基因序列,一段使蛋白质分泌到细胞外的信号肽序列,一段任意目标外源蛋白的完整DNA序列和一段保护mRNA稳定性的polyA序列。
进一步的,所述补全卵清蛋白基因第一内含子序列补全后将不再含有步骤S1中所选用的gRNA识别和切割的位点。
进一步的,所述一段补全卵清蛋白基因第一内含子序列,是从所选gRNA切割位点开始到第一内含子结束的序列或与切割位点上游序列一起构成完整的卵清蛋白基因第一内含子序列;所述信号肽序列为目标外源蛋白的原信号肽序列、鸡源溶菌酶信号肽、鸡源卵转铁球蛋白信号肽、鸡源类卵粘球蛋白信号肽或人工设计的信号肽;所述任意目标外源蛋白为人血清白蛋白、白细胞介素、人凝血因子、干扰素、肿瘤坏死因子、集落刺激因子、生长因子、趋化性细胞因子、重组抗体或者其他蛋白分子;所述polyA序列为利用鸡卵清蛋白原有polyA序列、额外添加BGH polyA序列、SV40polyA序列或者其他polyA序列。
进一步的,步骤S4包括以下步骤:
将步骤S2中制备好的gRNA/Cas9和步骤S3中制备好的ssDNA供体载体混合均匀后,与核酸转染试剂和精子孵育后通过人工授精转入母鸡个体,随后孵出鸡蛋,孵化直至出雏。
进一步的,所述S4步骤也可以通过以下方法实现:将步骤S2中制备好的任意gRNA/Cas9组合,和步骤S3中制备好的供体载体混合均匀后,通过电穿孔或者核酸转染试剂导入到从鸡胚中分离纯化得到的SSCs细胞中,通过药物筛选外源基因成功导入基因组的SSCs细胞,随后将筛选的PGCs发育成精子后,人工受精于母鸡个体。
本发明的有益效果是:
(1)本发明克服了鸡输卵管生物反应器的制备过程中转基因鸡的制备过程中基因随机整合的不确定性和低效率,实现外源基因特异性表达,相比较传统制备方法,操作更加简便,周期更短,效率更高。
(2)本发明利用CRISPR基因编辑技术和精子载体法两种技术,会弥补制备转基因动物时精子载体法外源DNA整合困难的缺陷,将两个技术有机结合建立一个低成本高效率的鸡遗传修饰的技术方法,提高了外源基因整合到基因组中的准确率,实现高效率导入外源基因,制备出能够表达并分泌外源蛋白到鸡蛋蛋清中的转基因鸡,并实现外源基因稳定的遗传给后代,且成本低廉,对卵原核无损伤。
附图说明
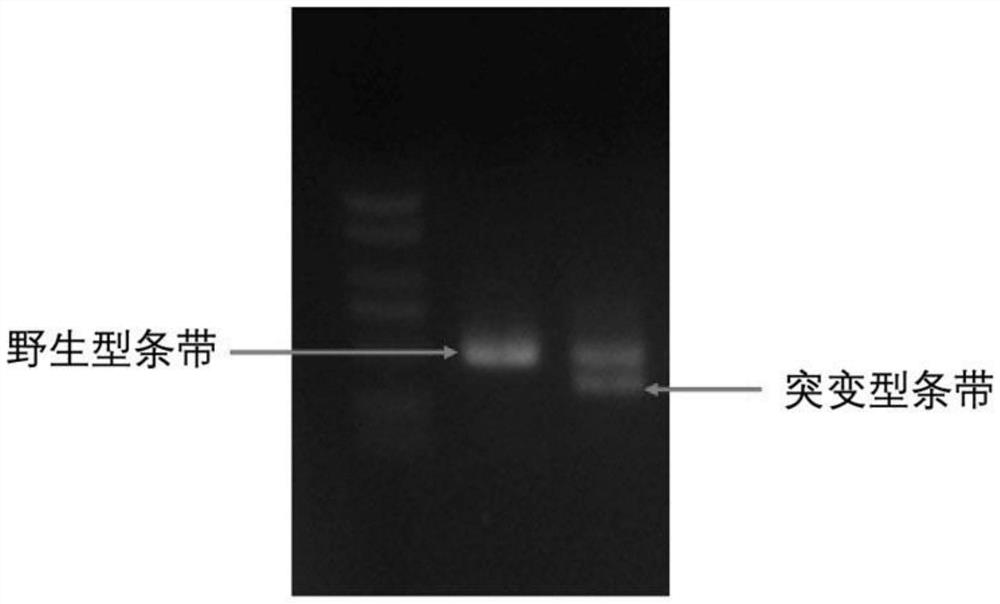
图1为本发明实施例1中T7E1酶切检测gRNA切割效率结果。
具体实施方式
下面结合附图进一步详细描述本发明的技术方案,但本发明的保护范围不局限于以下所述。
一种由精液介导的转基因鸡输卵管生物反应器的制备方法,包括以下步骤:
S1、设计靶向鸡基因组DNA的gRNA:选用鸡基因组目标DNA序列为卵清蛋白,选择同源性90%以上的gRNA,所述目标DNA的序列为:
5’-GTTTAAGTATCAGTAATTGGCTACCATTAACAACTGGCTCCTGAGAGGTCTTAAATGTAGAGACAGCTTTAAACTCAAAAGCACAGAGTGATTTTTAGAATAGATTTCCCAAGCAAAGAAAATAAACAGGGAGGAGCTTTAAGGGAGTAGCCATCTCATTATTATTATTATTTAAAGAAATGGCAGCAAGCCTACAAAAGAAAAATAAGACAGAGCAGAGAAGAAAGAGTCATGGTATGCTTTTCTATCTTAGCAAAATTAATCTCTACATGCCTAGGAAAAAGCCATGACAAGAGCAATCAGTTCAAAAGGTGTATGCAAAAAACCACATAATAGTAACTAGTACTGCATTGCCAGGAAGGAAGTTATGTCGCCATTCCATGGATCTCATTCTCATTTCCTTGCAGCTTGAGAGTATAATCAACTTTGAAAAACTGACTGAATGGACCAGTTCTAATGTTATGGAAGAGAGGAAGATCAAAGTGTACTTACCTCGCATGAAGATGGAGGAAAAATACAACCTCACATCTGTCTTAATGGCTATGGGCATTACTGACGTGTTTAGCTCTTCAGCCAATCTGTCTGGCATCTCCTCAGCAGAGAGCCTGAAGATATCTCAAGCTGTCCATGCAGCACATGCAGAAATCAATGAAGCAGGCAGAGAGGTGGTAGGGTCAGCAGAGGCTGGAGTGGATGCTGCAAGCGTCTCTGAAGAATTTAGGGCTGACCATCCATTCCTCTTCTGTATCAAGCACATCGCAACCAACGCCGTTCTCTTCTTTGGCAGATGTGTTTCCCCTAAAGAAGAAAGCTGAAAAACTCTGTCCCTTCCAACAAGACCCAGAGCACTGTAGTATCAGGGGTAAAATGAAAAGTATGTTCTCTGCTGCATCCAGACTTCATAAAAGCTGGAGCTTAATCTAGAAAAAAAATCAGAAAGAAATTACACTGTGAGAACAGGTGCAATTCACTTTTCCTTTACACAGAGTAATACTGGTAACTCATGGATGAAGGCTTAAGGGAATGAAATTGGACTCACAGTACTGAGTCATCACACTGAAAAATGCAACCTGATACATCAGCAGAAGGTTTATGGGGGAAAAATGCAGCCTTCCAATTAAGCCAGATATCTGTATGACCAAGCTGCTCCAGAATTAGTCACTCAAAATCTCTCAGATTAAATTATCAACTGTCACCAACCATTCCTATGCTGACAAGGCAATTGCTTGTTCTCTGTGTTCCTGATACTACAAGGCTCTTCCTGACTTCCTAAAGATGCATTATAAAAATCTTATAATTCACATTTCTCCCTAAACTTTGACTCAATCATGGTATGTTGGCAAATATGGTATATTACTATTCAAATTGTTTTCCTTGTACCCATATGTAATGGGTCTTGTGAATGTGCTCTTTTGTTCCTTTAATCATAATAAAAACATGTTTAAGCAAACACTTTTCACTTGTAGTATTTGAAGTACAGCAAGGTTGTGTAGCAGGGAAAGAATGACATGCAGAGGAATAAGTATGGACACACAGGCTAGCAGCGACTGTAGAACAAGTACTAATGGGTGAGAAGTTGAACAAGAGTCCCCTACAGCAA-3’;
S2、构建gRNA的表达载体:根据PAM规则确定靶位点序列后,合成符合表达载体构建要求的靶位点序列正负DNA单链,退火形成DNA双链之后连接到CRISPR/Cas系统表达载体上,载体导入细胞后能表达gRNA和Cas蛋白;PAM类型包括5'-NNN-3'、5'-NNNN-3'、5'-NNNNN-3'、5'-NNNNNN-3'、5'-NNNNNNN-3'和5'-NNNNNNNN-3';
S3、构建携带有外源基因表达框的供体载体,将供体载体转变为ssDNA:以双链DNA作为同源重组修复模板,将目标DNA位点DSB处上游和下游各100bp-1000bp序列,分别连接在外源基因的5'端和3'端,作为同源臂,供体载体ssDNA的形式存在;所述外源基因表达框包括一段补全卵清蛋白基因序列,一段使蛋白质分泌到细胞外的信号肽序列,一段任意目标外源蛋白的完整DNA序列和一段保护mRNA稳定性的polyA序列;所述补全卵清蛋白基因第一内含子序列补全后将不再含有步骤S1中所选用的gRNA识别和切割的位点;所述一段补全卵清蛋白基因第一内含子序列,是从所选gRNA切割位点开始到第一内含子结束的序列或与切割位点上游序列一起构成完整的卵清蛋白基因第一内含子序列;所述信号肽序列为目标外源蛋白的原信号肽序列、鸡源溶菌酶信号肽、鸡源卵转铁球蛋白信号肽、鸡源类卵粘球蛋白信号肽或人工设计的信号肽;所述任意目标外源蛋白为人血清白蛋白、白细胞介素、人凝血因子、干扰素、肿瘤坏死因子、集落刺激因子、生长因子、趋化性细胞因子、重组抗体或者其他蛋白分子;所述polyA序列为利用鸡卵清蛋白原有polyA序列、额外添加BGH polyA序列、SV40polyA序列或者其他polyA序列;
S4、将载体通过精子载体法人工授精导入鸡个体:将步骤S2中制备好的任意gRNA/Cas9组合,和步骤S3中制备好的供体载体混合均匀后,与核酸转染试剂和精液混合孵育后人工授精于选择的母鸡中,收集受精蛋并检测干扰素基因的表达。
实施例1
所述PAM种类为5'-NNN-3'时,用来源于Streptococcus pyogenes的SpCas9,PAM规则为5'-NGG-3',SpCas9在目标DNA序列中产生65条靶位点序列,如:
以pX330-U6-Chimeric_BB-CBh-hSpCas9质粒为载体(Addgene plasmid ID:42230,以下简称pX330),能够同时表达gRNA与SpCas9蛋白,将1μg pX330质粒用1μL BbsI酶切,37℃孵化1小时后,1%琼脂糖电泳回收酶切片段(QIAquick Gel Extraction Kit回收试剂盒),酶切反应体系如下:
根据选定的靶位点序列,按照规则人工合成两条寡核苷酸,规则如下:
其中,当靶位点序列的5'端不是碱基G时,需要额外加上碱基G;本例中需要合成的序列为5'-CACCGACCAACGCCGTTCTCTTCTT-3'和5'-AAACAAGAAGAGAACGGCGTTGGTC-3',将两条寡核苷酸链退火,形成短双链DNA,反应体系如下:
将上述反应体系在ep管中混合均匀,37℃放置1h后,95℃加热5min,然后放置在室温环境下冷却,退火过的寡核苷酸以1:200稀释到无菌水或EB中;
将经过BbsI酶切并纯化回收的线型pX330质粒与带有粘性末端的双链短DNA产物混合,LigaFastTM Rapid DNA Ligation System(Promega,Cat.No.M8221)进行连接反应,室温下放置30min,反应体系如下:
质粒转化:将DH5α感受态细胞从-80℃冰箱取出,迅速至冰上融化,然后取30μL感受态细胞和11μL连接产物加入准备好的1.5mL EP管中,轻弹管底,避免用移液枪吹打,静置30min;提前打开水浴锅,并升温至42℃,将静置后的混合液于水浴锅中热激1min后,迅速置于冰上静置2min,切勿摇动离心管;在酒精灯旁边,向离心管中加入500μL不含抗生素的LB培养基,于37℃恒温摇床上200rpm复苏50min;复苏完成后,在超净工作台中取100μL感受态涂布于含抗生素的LB培养平板上;将平板正置于37℃恒温培养箱中,待菌液被完全吸收后,倒置平板并过夜培养;待平板上长出肉眼可见并密度适中的菌落后,在酒精灯旁挑取单个菌落于装有500μL含抗生素的LB培养基的1.5mL EP管中,于37℃恒温摇床上200rpm摇菌5-6h;摇菌后,吸取1μL菌液作为模板,进行PCR反应,产物电泳后观察条带是否正确;菌落PCR验证后,吸取适量菌液送测序,验证插入序列是否正确;将正确的单克隆菌液扩大培养,收集菌液,并在8000g条件下离心10min,弃上清;按照天根无内毒素质粒小提试剂盒说明书中操作步骤提取质粒,获得无内毒素质粒pX330-gRNA;
T7E1酶切检测gRNA效率:将购买获得的UMNSAH/DF-1细胞(ATCCRCRL12203TM)复苏,添加含有10%胎牛血清的DMEM培养液,在39℃,5%CO
PCR扩增程序:95℃预变性5min;95℃变性15s,60℃退火15s,72℃延伸60s,35个循环后,72℃延伸5min,最后4℃保温;
用试剂盒NucleoSpin Gel and PCR Clean-up(MACHREY-NAGEL)回收PCR产物,取200ng产物进行T7Endonuclease I(NEB)酶切,酶切体系如下:
将反应体系混合好加入PCR管中,设置PCR仪温度设置,温度设置如下:
95℃10min
95℃-85℃-2℃/second
85℃-25℃-0.1℃/second
在反应体系中加入1ul T7Endonuclease I酶,37℃反应1小时后,电泳检测DNA条带。T7EI酶是一种非配对内切酶,在gRNA引导下,CRISPR/Cas9切割DNA发生突变,突变型DNA可与野生型DNA片段退火产生非配对DNA片段,T7EI酶能将其剪切。若没有发生突变,而无法被T7EI剪切。附图1中可以看到T7EI酶处理后,有比对照更小的DNA条带。
实施例2
以人干扰素β1基因为外源基因,构建供体质粒。
构建同源修复供体质粒:同源供体DNA依次由上游同源臂、外源基因表达框和下游同源臂顺次连接。外源基因表达框中包括一段第一内含子序列,用以补全卵清蛋白基因第一内含子序列;P2A自剪切序列;人干扰素β1基因,去除原有信号肽,添加鸡溶菌酶信号肽。
使用单链DNA作为同源重组修复模板,基因组断裂位点处上游800bp作为上游同源臂,下游800bp作为下游同源臂,取反义链作为模板进行人工合成,操作示意图如图所示,具体DNA模板序列如下:
5'-GACCAACGCCGTTCTCTTCTTTGGGTTTAAGTATCAGTAATTGGCTACCATTAACAACTGGCTCCTGAGAGGTCTTAAATGTAGAGACAGCTTTAAACTCAAAAGCACAGAGTGATTTTTAGAATAGATTTCCCAAGCAAAGAAAATAAACAGGGAGGAGCTTTAAGGGAGTAGCCATCTCATTATTATTATTATTTAAAGAAATGGCAGCAAGCCTACAAAAGAAAAATAAGACAGAGCAGAGAAGAAAGAGTCATGGTATGCTTTTCTATCTTAGCAAAATTAATCTCTACATGCCTAGGAAAAAGCCATGACAAGAGCAATCAGTTCAAAAGGTGTATGCAAAAAACCACATAATAGTAACTAGTACTGCATTGCCAGGAAGGAAGTTATGTCGCCATTCCATGGATCTCATTCTCATTTCCTTGCAGCTTGAGAGTATAATCAACTTTGAAAAACTGACTGAATGGACCAGTTCTAATGTTATGGAAGAGAGGAAGATCAAAGTGTACTTACCTCGCATGAAGATGGAGGAAAAATACAACCTCACATCTGTCTTAATGGCTATGGGCATTACTGACGTGTTTAGCTCTTCAGCCAATCTGTCTGGCATCTCCTCAGCAGAGAGCCTGAAGATATCTCAAGCTGTCCATGCAGCACATGCAGAAATCAATGAAGCAGGCAGAGAGGTGGTAGGGTCAGCAGAGGCTGGAGTGGATGCTGCAAGCGTCTCTGAAGAATTTAGGGCTGACCATCCATTCCTCTTCTGTATCAAGCACATCGCAACCAACGCCGTTCTCTTCTTTGGCAGATGTGTTTCCCCTggctccggcgctactaacttcagcctgctgaagcaggctggcgacgtggaggagaaccctggacctGCCACCATGaggtctttgctaatcttggtgctttgcttcctgcccctggctgctctggggACCAACAAGTGTCTCCTCCAAATTGCTCTCCTGTTGTGCTTCTCCACTACAGCTCTTTCCATGAGCTACAACTTGCTTGGATTCCTACAAAGAAGCAGCAATTTTCAGTGTCAGAAGCTCCTGTGGCAATTGAATGGGAGGCTTGAATACTGCCTCAAGGACAGGATGAACTTTGACATCCCTGAGGAGATTAAGCAGCTGCAGCAGTTCCAGAAGGAGGACGCCGCATTGACCATCTATGAGATGCTCCAGAACATCTTTGCTATTTTCAGACAAGATTCATCTAGCACTGGCTGGAATGAGACTATTGTTGAGAACCTCCTGGCTAATGTCTATCATCAGATAAACCATCTGAAGACAGTCCTGGAAGAAAAACTGGAGAAAGAAGATTTCACCAGGGGAAAACTCATGAGCAGTCTGCACCTGAAAAGATATTATGGGAGGATTCTGCATTACCTGAAGGCCAAGGAGTACAGTCACTGTGCCTGGACCATAGTCAGAGTGGAAATCCTAAGGAACTTTTACTTCATTAACAGACTTACAGGTTACCTCCGAAACTGAAAAGAAGAAAGCTGAAAAACTCTGTCCCTTCCAACAAGACCCAGAGCACTGTAGTATCAGGGGTAAAATGAAAAGTATGTTCTCTGCTGCATCCAGACTTCATAAAAGCTGGAGCTTAATCTAGAAAAAAAATCAGAAAGAAATTACACTGTGAGAACAGGTGCAATTCACTTTTCCTTTACACAGAGTAATACTGGTAACTCATGGATGAAGGCTTAAGGGAATGAAATTGGACTCACAGTACTGAGTCATCACACTGAAAAATGCAACCTGATACATCAGCAGAAGGTTTATGGGGGAAAAATGCAGCCTTCCAATTAAGCCAGATATCTGTATGACCAAGCTGCTCCAGAATTAGTCACTCAAAATCTCTCAGATTAAATTATCAACTGTCACCAACCATTCCTATGCTGACAAGGCAATTGCTTGTTCTCTGTGTTCCTGATACTACAAGGCTCTTCCTGACTTCCTAAAGATGCATTATAAAAATCTTATAATTCACATTTCTCCCTAAACTTTGACTCAATCATGGTATGTTGGCAAATATGGTATATTACTATTCAAATTGTTTTCCTTGTACCCATATGTAATGGGTCTTGTGAATGTGCTCTTTTGTTCCTTTAATCATAATAAAAACATGTTTAAGCAAACACTTTTCACTTGTAGTATTTGAAGTACAGCAAGGTTGTGTAGCAGGGAAAGAATGACATGCAGAGGAATAAGTATGGACACACAGGCTAGCAGCGACTGTAGAACAAGTACTAATGGGTGAGAAGTTGAACAAGAGTCCCCTACAGCAACCAAAGAAGAGAACGGCGTTGGC-3’;
选择DNA模板序列进行人工合成,然后用Takara的Guide-it
先对三个片段进行重叠PCR。将左臂、靶区和右臂对应的每个片段结合~20pmol,每个交界处有15-20bp的重叠。通过这种方式,创建1-2kb大小的捐赠模板。使用正向和反向引物对供体模板生成PCR产物。注意其中一个引物(正向或反向)必须包含5'磷酸化,以便分别生成反义或正义ssDNA。由于不可能确定哪种引物组合将产生产量和质量最高的ssDNA,获得以下四个引物,以同时产生正义链和反义链,如下所示:
Primer 1:Standard forward primer(F Primer)
Primer 2:5’-phosphorylated reverse primer(5’-P R Primer)
Primer 3:5’-phosphorylated forward primer(5’-P F Primer)
Primer 4:Standard reverse primer(R Primer)
建立两个100μl的PCR体系,如下图所示:
程序设为:
30–40 cycles:
98℃ 10sec
55℃ 5sec
72℃ 5sec/kb*
4℃ hold
每个PCR取5μl产物用于琼脂糖凝胶电泳分析,以检查dsDNA的正确扩增。使用提供的NucleoSpin凝胶和PCR试剂盒柱状纯化dsDNA底物。用纳米滴分光光度计(Thermo FisherScientific)测量DNA浓度。
为了生成ssDNA,每个PCR产物(阳性对照反应,PCR A或PCR B)都要经过两个短而连续的strandase反应(strandase A和strandase B反应)。在将Strandase A酶放在冰里的同时,上下移液几次,以便在向反应中加入正确的酶量之前实现均匀再悬浮。请按照如下试剂加入顺序,在室温下制备反应。Strandase A反应体系如下:
Strandase A反应孵育如下:
37℃ 5min/kb*
80℃ 5min
4℃ until next step;
按如下方法建立Strandase B反应,确保反应和管(从步骤2)冷却前4℃添加Strandase混合B同时保持Strandase B酶在冰上,在室温下制备反应;Strandase B反应体系如下:
Strandase B孵育反应如下:
37℃ 5min/kb*
80℃ 5min
4℃ until next step
最后柱状纯化ssDNA以去除游离核苷酸,使用NucleoSpin凝胶和PCR试剂盒以及Buffer NTC(针对ssDNA的结合缓冲液)。每个ssDNA样品在1-2%的琼脂糖凝胶上跑3μl,包括100-150ng的dsDNA底物作为对照。其余样品测浓度后存于4℃待用。
实施例3
精子载体法
采集新鲜公鸡的精液移入1mL获能液HTF中,水浴孵育40min,进行获能反应。取200nL悬浮于lmL M199培养基,混匀洗涤精浆:2500rpm离心5min,弃上清,重复洗涤一次,分别加入1mL各自孵育液,混匀。将实施例1中获得的gRNA表达质粒px330-gRNA与实施例2中获得的供体ssDNA 按1:1混合后与核酸转染试剂Lipofectamine LTX混匀,孵育10min后,加入含精子的孵育液中。置于37℃、5%CO
人工授精和检测:每只公鸡对应10只母鸡输精,连续输精2天,收集受精蛋7天。收集刚产下种蛋,打开蛋壳,将蛋黄倒入培养皿,翻转找到胚盘,用手术剪轻轻剪下胚盘,用镊子将胚盘转移至一个新的1.5mL离心管中,用手术剪剪碎。向放有剪碎胚盘的离心管中加入300μL禽血裂解液,再加入300μL裂解结合液,用移液枪吸打至胚盘完全溶解,然后将液相移入硅胶膜吸附柱(微量)中,12000rpm离心1min;弃废液,加入700μL80%乙醇,12000rpm离心1min,弃废液,12000rpm空离心3min,换灭菌套管,开口挥发乙醇2分钟,加入37℃预热无菌水50μL,室温静置5min,12000r/min离心1min,收集DNA。以收集DNA为模板,用引物进行PCR检测外源DNA,同时设置对照组,阳性对照模板为pX330质粒,阴性对照模板为未精子孵育处理受精蛋胚盘DNA。
以上所述仅是本发明的优选实施方式,应当理解本发明并非局限于本文所披露的形式,不应看作是对其他实施例的排除,而可用于各种其他组合、修改和环境,并能够在本文所述构想范围内,通过上述教导或相关领域的技术或知识进行改动。而本领域人员所进行的改动和变化不脱离本发明的精神和范围,则都应在本发明所附权利要求的保护范围内。
- 一种农杆菌介导结合浸没式培养获得转基因植物的方法
- 一种转基因鸡输卵管生物反应器的制备方法
- 一种转基因鸡输卵管生物反应器的制备方法