一种杞蓉片中多种有效成分的含量测定方法
文献发布时间:2023-06-19 19:27:02
技术领域
本发明属于药品质量检测分析技术领域,具体涉及一种杞蓉片中多种有效成分的含量测定方法。
背景技术
失眠发病的人群也很广,不论健康者、男性和女性、老年人和青年人、城市人和乡村人均可发生,失眠已经成为一种现代病,在市场经济比较发达,工作节奏较快的地区,失眠健忘的患者所占的比重更大,但实际未引起全社会的足够重视。有调查显示,全球42.5%的人存在着不同程度的失眠问题。成年人的失眠患病率为57%,30-50岁的失眠人群占到51%,尤其以男性居多。失眠,中医称为不寐,指睡眠时间深度减少的病症,会出现睡不着、早醒、头脑不清楚、记忆力减退、健忘等情况,长期失眠,易患失眠抑郁症。中医认为,肾亏、肾虚不藏,阳浮于外,遗精、阳萎早泄,也是导致失眠的重要原因之一。
目前治疗失眠健忘的方法有服用西药、中药和中医针灸治疗,常用的西药如受体激动剂和具有催眠效果的抗抑郁药物类药物,这些药物虽然能够减轻一些症状,但很难达到彻底治愈的目的,且药物副作用大,对全身的调节效果差。中药的治疗主要是按照辨证施治的原则,以补肾、安神、养心为主,临床常用药有杞蓉片、柏子养心胶囊、活力苏口服液、安神养心丸等,其中杞蓉片为西安千禾药业股份有限公司生产,具有补肾固精,益智安神的功能,用于肾亏遗精,阳萎早泄,失眠健忘。杞蓉片是由枸杞子、肉苁蓉、锁阳、蛇床子、女贞子、五味子、金樱子、淫羊藿、菟丝子九味药物经加工而成薄膜衣片。该品种因其在肾亏、肾阳虚所致的失眠健忘疗效确切,在临床使用过程中深受患者和医务工作者的认可,但是现行国家药品标准WS
锁阳为传统名贵中药材,具有补肾阳,益精血,润肠通便的功效,用于肾阳不足,精血亏虚,腰膝痿软,阳痿滑精,肠燥便秘等;锁阳含有黄酮类、萜类、甾体类、有机酸类、糖和糖苷类、挥发性成分和鞣质等,对人体免疫功能、性功能、肠功能、肾上腺皮质分泌功能都具有很好的增强和促进作用。现代药理研究表明,锁阳具有抗疲劳、保肝护肝及增强组织耐缺氧能力、提高机体免疫力、清除自由基、抑制人体免疫缺陷病毒(HIV)、抑制血小板聚集和雌激素样等广泛的药理作用。淫羊藿,具有补肾阳、强筋骨、祛风湿功效,用于肾阳虚衰所致阳痿遗精、筋骨痿软、风湿痹痛、麻木拘挛,是临床常用中药。现代药理研究表明,淫羊藿具有促进成骨细胞生长,镇静,抗抑郁,增强免疫功能,抗氧化,抗衰老等多种药理作用。女贞子具有滋补肝肾、明目乌发的作用,主要用于治疗肝肾阴虚、眩晕耳鸣、骨蒸潮热等症状,为常用扶正固本的补阴药;化学成分研究显示有效成分主要有萜类、苯乙醇类、多糖类等,现代药理研究表明,女贞子具有抗氧化、抗衰老、抗肿瘤、降血脂、免疫调节、清除自由基等多种药理作用。
锁阳在杞蓉片处方中为君药,为传统名贵中药材,且方中用量最大;女贞子为臣药,用量较大,但市场药材普遍存在掺杂、混用、乱用等现象,尤其是在药品采购验收鉴别过程中,发现锁阳、女贞子、淫羊藿的混淆品比较多,很混乱,还有在正品中加入伪品的现象。此外,2020年版《中国药典》一部中收载的锁阳药材,没有含量测定项,难以全面反映其内在质量。且原有的质量标准并不能全面反映杞蓉片药品的质量情况,因此有必要继续完善该产品的检测方法,确保该产品的质量和临床疗效。在原质量标准WS
发明内容
为克服现有技术的不足,本发明提供了一种杞蓉片中多种有效成分的含量测定方法,该方法对处方中锁阳的有效成分没食子酸、原儿茶酸、儿茶素、根皮苷,女贞子中有效成分红景天苷,淫羊藿中有效成分淫羊藿苷,共计6种有效成分同时进行测定。该检测方法一方面能够弥补现有方法的不足,提供一种更为全面、有效的药物控制质量标准,进一步有效保障产品质量;另一方面能够实现6种有效成分的同时检测,既节约了检测成本,试剂使用量的减少又符合绿色环保的要求。
为实现上述发明目的,本发明是采用如下技术方案来实现的:
一种杞蓉片的含量测定方法,所述杞蓉片由以下九味药材制备而成:枸杞子、肉苁蓉、锁阳、蛇床子、女贞子、五味子、金樱子、淫羊藿、菟丝子;采用HPLC方法同时测定锁阳的有效成分没食子酸、原儿茶酸、儿茶素、根皮苷,女贞子中有效成分红景天苷,淫羊藿中有效成分淫羊藿苷,其包含以下步骤:
1)色谱条件与系统适用性试验
以十八烷基硅烷键合硅胶为填充剂;以有机溶剂为A相、一定浓度的稀酸水溶液为B相,梯度洗脱,所述有机溶剂为选自甲醇、乙腈或乙腈-甲醇混合液,所述稀酸水溶液选自重量或体积百分浓度0.01%-1.0%的磷酸、甲酸、冰乙酸或以上三种的混合;柱温10-40℃;二极管阵列检测器(PDA),测定没食子酸、原儿茶酸、儿茶素、根皮苷检测波长275-285nm;测定红景天苷检测波长265-275nm;测定淫羊藿苷检测波长270-280nm;流速为0.8ml/min-1.2ml/min;进样体积为10-20μl。
2)对照品溶液的制备
精密称取各对照品适量,分别加甲醇制成没食子酸0.1mg/ml、原儿茶酸0.06mg/ml、儿茶素0.14mg/ml、根皮苷0.12mg/ml、红景天苷0.06mg/ml、淫羊藿苷0.2mg/ml贮备液;依次精密吸取上述6种贮备液各1ml置于同一10ml棕色容量瓶中,加甲醇溶解并定容,摇匀,作为质量浓度分别为10,6,14,12,6,20μg/ml的混合对照品溶液。
3)供试品溶液的制备
取杞蓉片20片,除去薄膜衣,精密称定,研细,混匀,取约0.2-1.0g,精密称定,置具塞锥形瓶中,精密加入甲醇20-50ml,称定重量,超声处理30-50min(250W,50kHz),放冷,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
4)测定
分别精密吸取对照品溶液与供试品溶液各10-20μl,注入高效液相色谱仪,测定,即得测定结果。
优选的,所述A相为乙腈,B相为体积百分浓度0.05%的磷酸水溶液;流速为1.0ml/min;柱温为30℃;测定没食子酸、原儿茶酸、儿茶素、根皮苷检测波长为280nm;测定红景天苷、淫羊藿苷的检测波长为273nm。
优选的,梯度洗脱时,梯度洗脱程序为:0~10min,8%A;10~20min,8~20%A;20~30min,20%~35%A;35~35min,35~45%A;35~45min,8%A;
具体梯度洗脱程序为:
优选的,取杞蓉片20片,除去薄膜衣,精密称定,研细,混匀,称取0.4g,精密称定,置具塞锥形瓶中,精密加入甲醇25ml,称定重量,超声处理40min(250W,50kHz),放冷,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
本发明中杞蓉片具体处方为:枸杞子70g、肉苁蓉140g、锁阳210g、蛇床子42g、女贞子56g、五味子42g、金樱子56g、淫羊藿98g、菟丝子84g。
与现有技术相比,本发明具有以下有益的技术效果:
本发明杞蓉片的含量测定方法,在原质量标准WS
附图说明
下面结合附图对本发明作进一步的说明:
图1为本发明专属性试验的空白试剂色谱图。
图2为本发明专属性试验的缺锁阳阴性样品色谱图。
图3为本发明专属性试验的缺女贞子阴性样品色谱图。
图4为本发明专属性试验的缺淫羊藿阴性样品色谱图。
图5为本发明在色谱条件下对照品色谱图。
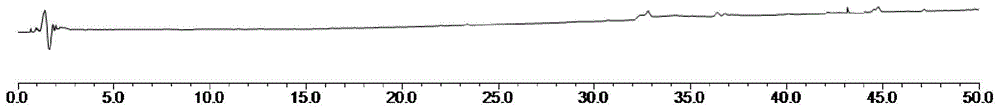
图6为本发明在色谱条件下供试品色谱图;其中,1为没食子酸峰、2为原儿茶酸峰、3为儿茶素峰、4为根皮苷峰、5为红景天苷峰、6为淫羊藿苷峰。
图7为本发明中6种成分的线性关系图。
具体实施方式
以下参照具体的实施例来说明本发明。本领域技术人员能够理解,这些实施例仅用于说明本发明,其不以任何方式限制本发明的范围。下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的原辅料、试剂材料等,如无特殊说明,均为市售购买产品。
实施例1:杞蓉片的处方及制备方法
处方:枸杞子70g、肉苁蓉140g、锁阳210g、蛇床子42g、女贞子56g、五味子42g、金樱子56g、淫羊藿98g、菟丝子84g
制法:以上九味,锁阳粉碎成细粉,过筛;五味子、蛇床子用60%乙醇回流提取,第一次2小时,第二次1小时,合并提取液,滤过,滤液回收乙醇,浓缩至适量;其余枸杞子等六味加水煎煮二次,第一次3小时,第二次2小时;合并煎液,静置,取上清液浓缩至适量,加入上述五味子清膏充分搅匀,浓缩成稠膏,加入锁阳细粉混匀,干燥,粉碎,加入淀粉、羟丙纤维素、二氧化硅适量,制粒,压制成1000片,包薄膜衣,即得。
实施例2:检测波长的选择
通过对没食子酸、原儿茶酸、儿茶素、根皮苷、红景天苷、淫羊藿苷6种对照品溶液的紫外吸收光谱,并参考《中国药典》2020年版一部及相关文献资料可知,没食子酸、原儿茶酸在280nm有最大吸收,儿茶素在210nm和280nm有最大吸收,根皮苷在225nm和285nm有最大吸收,红景天苷在224nm和275nm有最大吸收,淫羊藿苷在270nm有最大吸收;虽然儿茶素在210nm、根皮苷在225nm、红景天苷在224nm低波段也有最大吸收,但色谱背景的干扰也大,因没食子酸、原儿茶酸、儿茶素、根皮苷高波段的最大吸收波长相近,综合考虑分析后,最终将没食子酸、原儿茶酸、儿茶素、根皮苷的检测波长确定为280nm;同理将红景天苷和淫羊藿苷的检测波长确定为273nm。
试验表明,采用二极管阵列检测器(PDA)波长切换技术进行检测,用二极管阵列检测器综合考虑不同有效成分最强吸收且杂质干扰较小的波长进行波长切换检测。可在一张色谱图中较好的对测定组分进行分离;去除干扰杂质,有利于组分含量准确计算。
实施例3:流动相的选择
为使供试品中6种有效成分能得到有效的分离,同时峰形、理论塔板数等达到要求,预试验分别采用乙腈-0.5%冰乙酸溶液、甲醇-0.5%磷酸溶液、乙腈-0.1%磷酸溶液、甲醇-乙腈-0.2%磷酸溶液、乙腈-0.05%磷酸溶液等不同比例的流动性进行试验。比如,采用流动相乙腈-0.5%冰乙酸溶液等度洗脱比例20:60或60:40时,前者保留时间过长,后者部分主峰与杂质未能有效分离且峰形不好。采用流动相甲醇-0.5%磷酸溶液等度洗脱比例20:80或60:40时,前者30分钟内未出峰且基线漂移,后者部分主峰与杂质未能有效分离且峰形不好。采用流动相乙腈-0.1%磷酸溶液等度洗脱比例20:80或60:40时,前者30分钟内未出峰且基线漂移,后者部分主峰与杂质未能有效分离且峰形不好。采用流动相甲醇-乙腈-0.2%磷酸溶液等度洗脱比例10:10:80或30:30:40时,有效成分峰有拖尾现象,但前者保留时间过长,后者主峰与杂质未能有效分离。采用流动相乙腈-0.05%磷酸溶液等度洗脱比例25:75或75:25时,基线平稳,保留时间长,但红景天苷峰有重叠。结果表明,以乙腈-0.05%磷酸水溶液线性梯度分离效果最好,基线最平稳,但保留时间较长,故最终选择流动相A相为乙腈,流动相B相为体积百分浓度0.05%的磷酸水溶液进行梯度洗脱试验,经过多次试验都不理想,比如,采用流动相乙腈-0.05%磷酸溶液梯度洗脱,0~8min,A:8;8~20min,A:8→20;20~35min,A:20→35;35~45min,A:35→45;45~50min,A:8;结果没食子酸峰、原儿茶酸峰未完全分开。采用流动相乙腈-0.05%磷酸溶液梯度洗脱,0~10min,A:8;10~30min,A:8→20;30~35min,A:20→35;35~45min,A:35→45;45~50min,A:8;结果儿茶素峰、红景天苷峰未完全分开,其余4种有效成分的分离度、峰形、理论塔板数等符合要求。经最终试验发现,采用流动相乙腈-0.05%磷酸溶液梯度洗脱,梯度洗脱程序为:0~10min,8%A;10~20min,8~20%A;20~30min,20%~35%A;35~35min,35~45%A;35~45min,8%A;结果在进行梯度洗脱的条件下分离效果最好,基线平稳,6种有效成分成分的色谱峰可完全分离,各组分出峰时间、峰型和分离度均符合要求,故该条件能满足分析测定的相关要求。
实施例4:专属性研究
4.1各阴性样品的制备:
按样品的处方工艺和配方比例(实施例1),分别制备不含锁阳、女贞子和淫羊藿的阴性样品;如不含锁阳阴性样品的制备:五味子、蛇床子用60%乙醇回流提取,第一次2小时,第二次1小时,合并提取液,滤过,滤液回收乙醇,浓缩至适量;枸杞子、肉苁蓉、女贞子、金樱子、淫羊藿、菟丝子六味加水煎煮二次,第一次3小时,第二次2小时;合并煎液,静置,取上清液浓缩至适量,加入上述五味子清膏充分搅匀,浓缩成稠膏,加入淀粉、羟丙纤维素、二氧化硅适量,搅匀,真空干燥,粉碎成细粉,即得。
同法制备不含女贞子和淫羊藿的阴性样品。
4.2阴性样品溶液的制备:
分别称取各阴性样品0.4g,精密称定,置具塞锥形瓶中,精密加入甲醇25ml,称定重量,超声处理50min(250W,50kHz),放冷,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
4.3供试品溶液的制备:
试验方法:取同一批号杞蓉片20片,除去薄膜衣,精密称定,研细,混匀,取约0.2-1.0g,精密称定,置具塞锥形瓶中,精密加入甲醇20-50ml,称定重量,超声处理30-90min(250W,50kHz),放冷,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。分别精密吸取对照品溶液与供试品溶液各10μl,注入高效液相色谱仪,测定,即得测定结果。
方法1:精密加甲醇25ml,精密称定,超声提取30min,放冷补足重量,过滤;
方法2:精密加80%甲醇25ml,精密称定,超声提取30min,放冷补足重量,过滤;
方法3:精密加60%甲醇25ml,精密称定,超声提取30min,放冷补足重量,过滤;
方法4:精密加甲醇25ml,精密称定,加热回流提取60min,放冷补足重量,过滤;
方法5:精密加80%甲醇25ml,精密称定,加热回流提取60min,放冷补足重量,过滤;
方法6:精密加60%甲醇25ml,精密称定,加热回流提取90min,放冷补足重量,过滤;
方法7:精密加甲醇25ml,精密称定,超声提取35min,放冷补足重量,过滤;
方法8:精密加甲醇25ml,精密称定,超声提取40min,放冷补足重量,过滤;
方法9:精密加甲醇25ml,精密称定,超声提取50min,放冷补足重量,过滤;
表1供试品制备方法的选择试验结果
表1结果表明:采用方法1时各有效成分能得到有效提取,但峰面积略低于方法8,表明超声提取30min有效成分未能100%转移;采用方法2和方法3时因甲醇浓度较低各有效成分峰面积明显较低,说明有效成分转移率较低;采用方法4、方法5和方法6时,原儿茶酸峰、红景天苷峰面积较小,可能因黄酮类成分在受热过程中存在损失;采用方法7时红景天苷提取不充分,峰面积明显低于方法8和方法9时;采用方法8和方法9时的峰面积无显著性差异,但方法9超声提取时间较长;故确定最佳的供试品溶液的制备方法为方法8:杞蓉片20片,除去薄膜衣,精密称定,研细,混匀,称取0.4g,精密称定,置具塞锥形瓶中,精密加入甲醇25ml,称定重量,超声处理40min(250W,50kHz),放冷,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
4.4色谱条件与系统适用性试验
以以十八烷基硅烷键合硅胶为填充剂(250mm×4.6mm,5μm)的色谱柱;按表1中确定的流动相A相为乙腈,B相为体积百分浓度0.05%的磷酸水溶液;流速为1.0ml/min;柱温为30℃;测定没食子酸、原儿茶酸、儿茶素、根皮苷检测波长为280nm;测定红景天苷、淫羊藿苷的检测波长为273nm;进样体积为10μl;理论板数均应不低于3000。
4.5对照品溶液的制备:
精密称取各对照品适量,分别加甲醇制成没食子酸0.1056mg/ml、原儿茶酸0.0631mg/ml、儿茶素0.1452mg/ml、根皮苷0.1265mg/ml、红景天苷0.0586mg/ml、淫羊藿苷0.2104mg/ml贮备液;依次精密吸取上述6种贮备液各1ml置于同一10ml棕色容量瓶中,加甲醇溶解并定容,摇匀,作为质量浓度分别为10.56,6.31,14.52,12.65,5.86,21.04μg/ml的混合对照品溶液。
4.6测定:
分别精密吸取各阴性样品溶液、混合对照品溶液、供试品溶液各10μl,注入LC-2030CPlus型高效液相色谱仪(PDA检测器,岛津),测定。
结果显示:供试品溶液色谱图中,在6种对照品色谱峰相应的位置上有相同的色谱峰;阴性无干扰,且分离度和理论板数均符合要求;表明该色谱条件专属性良好。具体见下附图4。
实施例5:重复性试验
5.1色谱条件与系统适用性试验同实施例4
5.2精密吸取同一供试品溶液10μl,注入液相色谱仪,重复进样6次,计算峰面积RSD。
5.3结果显示,6种有效成分峰面积的RSD值分别为0.71%、1.15%、1.11%、0.86%、1.07%、0.93%,均小于2.0%;表明精密度良好。具体结果见下表2。
表2:重复性实验6种有效成分峰面积数据表
实施例6:线性范围
6.1色谱条件与系统适用性试验同实施例4
6.2分别精密吸取各对照品贮备液1.5ml置于10ml棕色量瓶中用甲醇定容,得混合对照品溶液,再分别精密吸取上述混合对照品溶液各2μl、5μl、8μl、10μl、15μl、20μl、按实施例3所述色谱条件进行检测,记录色谱图,以峰面积(Y)对浓度(X)进行线性回归分析,绘制线性关系图,报告线性方程。结果见表3和图7。
表3:6种成分的标准曲线方程和相关系数及线性范围
实施例7:准确度(回收率)
7.1色谱条件与系统适用性试验同实施例4
7.2供试品溶液的制备:
取杞蓉片20片,除去薄膜衣,精密称定,研细,混匀,称取0.4g样品各6份,精密称定,置10ml棕色量瓶中,再分别精密吸取没食子酸0.1056mg/ml、原儿茶酸0.0631mg/ml、儿茶素0.1452mg/ml、根皮苷0.1265mg/ml、红景天苷0.0586mg/ml、淫羊藿苷0.2104mg/ml的贮备液各1ml置于上述棕色量瓶中加甲醇适量,超声处理(250W,40kHz)40min,放冷,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。测定:分别精密吸取对照品溶液、供试品溶液各10μl,注入高效液相色谱仪,测得没食子酸、原儿茶酸、儿茶素、根皮苷、红景天苷、淫羊藿苷的回收率和RSD%。
表4:回收率试验数据表
回收率%=(C-A)/B×100%,式中A为供试品所含被测成分量;B为加入对照品量;C为实测值。
结果见表4,标准要求:回收率要求85%~110%,回收率RSD<4.0%;实验结果表明该方法准确度良好。
实施例8:中间精密度
8.1色谱条件与系统适用性试验同实施例4
8.2在同一实验室,由不同实验人员A和B在不同日期,使用不同色谱仪、进行实验,计算各有效成分含量。结果见表5,结果表明发明专利方法精密度良好。
表5:中间精密度试验数据表
实施例9:耐用性
9.1色谱条件与系统适用性试验同实施例4
9.2取同一供试品溶液,室温下放置,分别于0、4、8、12、24小时,精密吸取10μl注入高效液相色谱仪;结果见表6,结果表明,供试品溶液24小时内保峰面积的RSD最大值仅为0.19%,远小于标准2.0%;表明试品溶液在室温条件下24h内稳定。
表6:稳定性试验数据表
综上所述:本发明杞蓉片中多种有效成分的含量测定方法,从专属性、重复、准确度(回收率)、中间精密度、耐用性等方面验证了本方法的科学性。通过对处方中3味药材中6种有效成分含量的测定,进一步提升了产品质量标准,保证了杞蓉片的安全性和有效性,弥补了现有技术的不足。
实施例10:稳定性试验
10.1色谱条件与系统适用性试验同实施例4
10.2取杞蓉片市售样品(批号:20200301、20200302、20200303,西安千禾药业股份有限公司生产)进行加速稳定性实验和室温长期稳定性实验。试验结果见表7。
表7:杞蓉片加速及长期稳定性试验数据
加速实验条件:将样品放置在相对湿度为75%±5%,温度为40℃±2℃的稳定性试验箱中进行6个月加速实验考察,分别在第0、1、2、3、6月取样进行检测;室温实验条件:将样品放置在相对湿度60%±5%,温度25℃±2℃的稳定性试验箱中进行长期24个月的实验考察,分别在第0、3、6、12、18月、24个月检查。
实验结果表明,加速试验6个月和长期实验24个月,所检测的有效成分没食子酸、原儿茶酸、儿茶素、根皮苷、红景天苷、淫羊藿苷含量均未发生显著性变化,符合要求,说明该检测方法,能有效控制药物的质量,可考虑纳入质量标准中。另,从连续三批售产品(批号:20200301、20200302、20200303)24个月末的检测结果来看,没食子酸的平均含量为0.199mg/片、原儿茶酸的平均含量为0.114mg/片、儿茶素的平均含量为0.286mg/片、根皮苷的平均含量为0.112mg/片、红景天苷的平均含量为0.100mg/片、淫羊藿苷的平均含量为0.415mg/片;考虑到不同原料之间的差异及生产及检验过程种的误差,建议可将含量标准定为:含没食子酸≥0.18mg/片、原儿茶酸≥0.10mg/片、儿茶素≥0.27mg/片、根皮苷≥0.10mg/片、红景天苷≥0.09mg/片、淫羊藿苷≥0.40mg/片。
以上给出的实施例是实现本发明较优的例子,本发明不限于上述实施例。本领域的技术人员根据本发明技术方案的技术特征所做出的任何非本质的添加、替换,均属于本发明的保护范围。
- 一种荷丹片中多种有效成分的含量测定方法
- 一种荷丹片中多种有效成分的含量测定方法