公猪精子活力miRNA标志物及其应用
文献发布时间:2023-06-19 19:27:02
技术领域
本发明属于生物技术领域,尤其涉及公猪精子活力miRNA标志物及其应用。
背景技术
公猪精子活力是精子运动参数指标,精子通过在附睾、前列腺等副性腺中不断发育逐渐具备向前游动的能力。精浆外泌体来源于雄性生殖道中,是构成精子周围环境的重要部分,可以通过蛋白质、miRNA等分子运送至精子进行物质交换和信息调节,完善精子的结构与功能,对精子活力起到重要的调节作用。精浆外泌体中含有大量的miRNA,研究发现活力不同的精液在一些物种中均存在miRNA表达差异,精子活力异常可能是由于发育过程中受到这些miRNA表达失调影响导致精子发育不全。
miRNA可以通过与AGO家族蛋白形成复合物的方式沉默mRNA,调控mRNA的稳定性和蛋白质的合称,最终影响到精子的基本参数以及受精过程中的各个环节。分泌外泌体的源细胞的功能状态的不同能够通过其分泌外泌体的miRNA种类、含量得到体现,因此精浆外泌体中miRNA在成为精子活力的无创标志物进而预测公猪繁殖力中拥有非常广阔的发展空间。
虽然对于总体上精浆外泌体已有一定的研究成果,但其在猪这一物种中的研究仍然较为匮乏。有研究对猪精浆外泌体中的miRNA进行鉴定分析([1]吴志胜,陈慧芳,刘俊杰,等.长白猪精浆外泌体miRNAs的鉴定与功能分析[J].农业生物技术学报,2021.),发现一些高丰度miRNA对精子功能具有潜在的调控作用,但其并未揭示不同miRNA在不同活力猪精液精浆外泌体中的表达差异,因此目前对不同miRNA在不同活力猪精液精浆外泌体中表达差异的研究依旧存在空白。
发明内容
基于此,本发明提供了一种公猪精子活力miRNA标志物及其应用,其对猪精子活力的鉴定具有可靠性、准确性。
本发明的第一方面,提供公猪精子活力miRNA标志物,其特征在于,包括:ssc-miR-486、ssc-miR-10386、ssc-miR-708-5p、ssc-miR-122-5p、ssc-miR-199a-3p和ssc-miR-31。
在其中一些实施例中,所述ssc-miR-486、ssc-miR-10386、ssc-miR-708-5p、ssc-miR-122-5p和ssc-miR-199a-3p在低活力公猪精液精浆外泌体中高表达,在高活力公猪精液精浆外泌体中低表达,所述ssc-miR-31在低活力公猪精液精浆外泌体中低表达,在高活力公猪精液精浆外泌体中高表达,所述高表达是指低活力公猪精液精浆外泌体中的表达量/高活力公猪精液精浆外泌体中的表达量>1.5,所述低表达是指低活力公猪精液精浆外泌体中的表达量/高活力公猪精液精浆外泌体中的表达量<0.85。
本发明提供了5种在低活力公猪精液精浆外泌体中高表达,在高活力公猪精液精浆外泌体中低表达的miRNA,另外还提供了一种在低活力公猪精液精浆外泌体中低表达,在高活力公猪精液精浆外泌体中高表达的miRNA,由于同时采用不同的miRNA作为标志物,且包含表达情况不同的miRNA标志物,提高了对猪精子活力鉴定的准确率,降低了假阳性的概率。
本发明的第二方面,要求保护上述标志物在制备鉴定公猪精子活力的试剂盒中的应用。
进一步的,所述ssc-miR-486的核苷酸序列如SEQ ID NO.1所示,所述ssc-miR-10386的核苷酸序列如SEQ ID NO.2所示,所述ssc-miR-708-5p的核苷酸序列如SEQ IDNO.3所示,所述ssc-miR-122-5p的核苷酸序列如SEQ ID NO.4所示,所述ssc-miR-199a-3p的核苷酸序列如SEQ ID NO.5所示,所述ssc-miR-31的核苷酸序列如SEQ ID NO.6所示。
本发明的第三方面,提供用于检测权利要求1所述的miRNA标志物在公猪精浆外泌体中表达量的引物,其特征在于,包括:
ssc-miR-486的上游引物,其核苷酸序列如SEQ ID NO.7所示;
ssc-miR-10386的上游引物,其核苷酸序列如SEQ ID NO.8所示;
ssc-miR-708-5p的上游引物,其核苷酸序列如SEQ ID NO.9所示;
ssc-miR-122-5p的上游引物,其核苷酸序列如SEQ ID NO.10所示;
ssc-miR-199a-3p的上游引物,其核苷酸序列如SEQ ID NO.11所示。
进一步的,还包括ssc-miR-31的上游引物,其核苷酸序列如SEQ ID NO.12所示。
本发明的第四方面,要求保护上述引物在制备鉴定公猪精子活力的试剂盒中的应用。
本发明的第五方面,提供一种用于鉴定公猪精子活力的试剂盒,所述试剂盒含有上述引物。
与现有技术相比,本发明的优点在于:
1、本发明的六个miRNA标志物在猪低活力精液和高活力精液中差异表达,既包含在低活力猪精液中上调表达的ssc-miR-486、ssc-miR-10386、ssc-miR-708-5p、ssc-miR-122-5p、ssc-miR-199a-3p,又包含在低活力精液中下调表达的ssc-miR-31,从而提高了对猪精液质量鉴定的可靠性、准确性。
2、对猪精浆外泌体的实时荧光定量PCR显示,6个差异表达miRNA标志物均具有一定的差异倍数,表明6个差异表达miRNA是鉴定猪精子活力的优良指标,将其用于鉴定猪精子活力具有良好前景。本发明一方面可用于猪精液的筛选,另一方面可以用于对公猪进行选育。
附图说明
图1为测序样品组间活力差异图;
图2为测序数据序列长度分布图;
图3为各组miRNA鉴定结果图;
图4为miRNA表达量概率分布图;
图5为差异表达miRNA火山图;
图6为在低活力组上调表达的已知miRNA热图;
图7为在低活力组下调表达的已知miRNA热图;
图8为生物过程中基因富集数目最多的前20位条目GO分析结果图;
图9为细胞成分中基因富集数目最多的前20位条目GO分析结果图;
图10为分子功能中基因富集数目最多的前20位条目GO分析结果图;
图11为差异表达miRNA靶基因KEGG富集分析结果图;
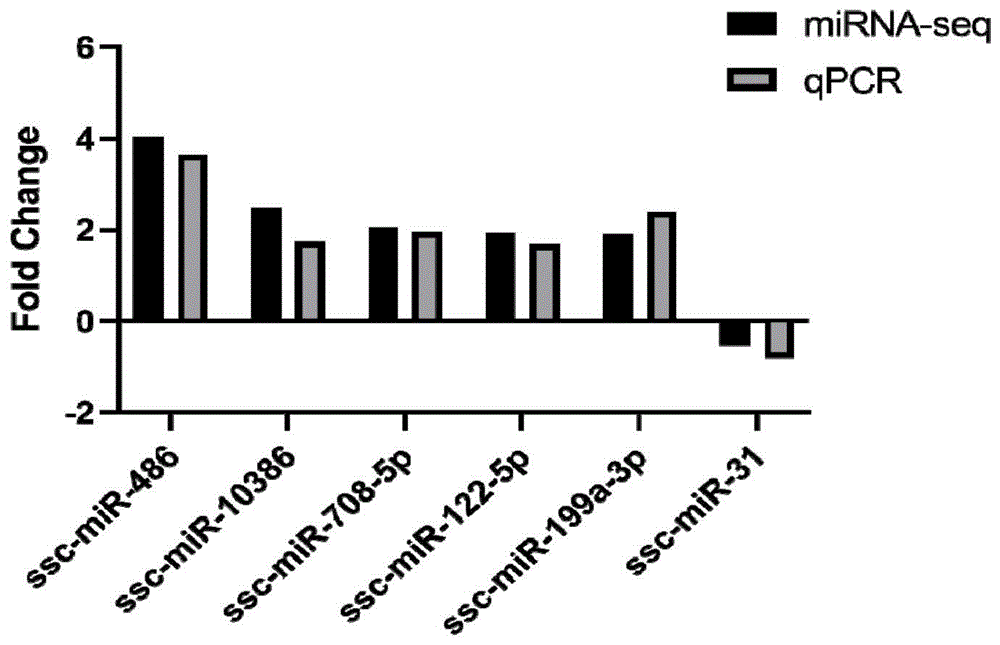
图12为差异表达miRNA验证结果图。
具体实施方式
为了便于理解本发明,下面将参照相关附图对本发明进行更全面的描述。附图中给出了本发明的较佳实施例。但是,本发明可以以许多不同的形式来实现,并不限于本文所描述的实施例。相反地,提供这些实施例的目的是使对本发明的公开内容的理解更加透彻全面。
除非另有定义,本文所使用的所有的技术和科学术语与术语本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在限制本发明。
本发明中,“第一方面”、“第二方面”、“第三方面”、“第四方面”等仅用于描述目的,不能理解为指示或暗示相对重要性或数量,也不能理解为隐含指明所指示的技术特征的重要性或数量。而且“第一”、“第二”、“第三”、“第四”等仅起到非穷举式的列举描述目的,应当理解并不构成对数量的封闭式限定。
本发明中涉及的百分比含量,如无特别说明,对于固液混合和固相-固相混合均指质量百分比,对于液相-液相混合指体积百分比。本发明中涉及的百分比浓度,如无特别说明,均指终浓度。所述终浓度,指添加成分在添加该成分后的体系中的占比。本发明中的温度参数,如无特别限定,既允许为恒温处理,也允许在一定温度区间内进行处理。所述的恒温处理允许温度在仪器控制的精度范围内进行波动。
在本发明的下述实施例中,所使用的实验仪器和试剂如下:
试验主要仪器如下:超速冷冻离心机,BeckMan,美国;冷冻离心机,Eppendorf,德国;17℃恒温箱,猪仙子,中国;2100生物分析仪系统,Agilent,美国;QuanStudio 3实时荧光定量PCR仪,Thermo Scientific,美国;PCR仪,BioRad,美国;CASA,Hamilton Thorne,美国。
试验主要试剂耗材如下:RNA提取试剂盒,Qiagen,美国;cDNA合成试剂盒,天根生物,中国;miRNA荧光定量检测试剂盒,天根生物,中国;引物,生工生物,中国;RNA Nano6000Assay Kit试剂盒,Agilent,美国;QIAseq miRNA Library Kit,Qiagen,美国;TruSeqPE-Cluster Kit v3-cBot-HS,Illumina,美国;0.45um滤器,Millipore,美国;0.22um滤器,Millipore,美国;PBS溶液,索莱宝,中国;超离管,BeckMan,美国。
实施例1、检测筛选不同精液活力精浆外泌体中的差异表达miRNA
1.1精浆样品的采集
6头15-25月龄长白公猪由北方某公猪站地区提供,所有公猪均按公猪站制定的标准方案进行饲养管理。使用手握法进行精液采集。精液样品采集后进行CASA检测,取近三月精子活力均高于90的长白公猪分别采精,要求采集当次精子活力高于90%,称为高活力组(High Motility Group)(三份精液样品编号依次为HM1、HM2、HM3);取近三月精子活力5次及以上低于80的长白公猪采精,要求采集当次精子活力低于80%,称为为低活力组(LowMotility Group)样本(三份精液样品编号依次为LM1、LM2、LM3)。
通过计算机辅助分析系统(CASA)对用于分离和测序的长白公猪精子活力进行检测(表1),畸形率均低于20%,符合国家制定的种猪常温精液质量标准。活力检测结果经独立t检验分析显示:两组间精子活力存在极显著统计学差异(P<0.001),如图1所示。
表1精液样品的CASA检测结果
1.2精浆外泌体分离
使用超速离心法分离提取精浆外泌体:
(1)在37℃条件下解冻精浆;
(2)4℃温度下,以3000g离心力离心30min,此时沉淀为细胞碎片,保留纯净的上清液于新管进行下一步操作;
(3)以12000g,4℃离心70min,分离出细胞外大囊泡,保留上清液;
(4)得到的样品液依次使用0.45μm、0.22μm针孔滤器过滤;
(5)将过滤后的液体使用超高速离心机以4℃,100000g的条件离心80min;
(6)去除上清液后加入PBS,反复吹打使外泌体沉淀重溶,此时能肉眼观察到白色胶絮状物质即为外泌体;
(7)重溶得到的液体使用超高速离心机以4℃,100000g条件再次离心80min以纯化外泌体;
(8)使用400ul PBS溶液重溶外泌体,为保证其活性存放于-80℃冰箱。
1.3外泌体总RNA提取
依据说明书步骤,使用miRNeasy Serum/Plasma Advanced Kit试剂盒对精浆外泌体总RNA进行提取,之后取部分RNA使用RNA Nano 6000Assay Kit试剂盒(Agilent)进行处理,处理后采用2100生物分析仪系统(Agilent)检测总RNA的浓度值和纯度值。总RNA的详细提取步骤如下:
(1)向100μl样品中加入700μl的QIAzol裂解液,涡旋混匀;
(2)15-25℃环境条件下匀浆5分钟;
(3)上述处理后样本每管中添加140μl的三氯甲烷,将管盖拧紧,大力晃动15s;
(4)室温下孵育5-10分钟;
(5)4℃条件下以12000g离心力离心15分钟;
(6)离心得到的上清液转移到新管中,吸取时避开下层液体,之后取上清液1.5倍体积的无水乙醇与其混匀;
(7)取体积为700μl的样本于RNeasy Mini柱中,置于收集管中。常温条件离心8000g,15秒。丢弃管中液体;
(8)重复步骤7,收集剩余的样本;
(9)取体积为700μl的Buffer RWT于柱内,使用8000g离心力离心15秒,丢弃多余的过滤液体;
(10)取体积为500μl的Buffer RPF于柱内,使用8000g离心力离心15秒,丢弃多余的过滤液体;
(11)取体积为500μl的Buffer RPE于柱内,使用8000g离心力离心15秒,丢弃多余的过滤液体;
(12)取RNeasy Mini柱置入全新收集管中,调整离心机为12000g运行2分钟;
(13)取RNeasy Mini柱置入全新EP管中,打开管盖,常温环境下静置2分钟;
(14)向RNeasy Mini柱加入35ul RNA free water,盖上盖子室温静置2分钟;
(15)4℃使用12000g离心力离心2分钟;
(16)将离心下的液体中心加入RNeasy Mini柱,室盖上盖子室温静置2分钟;
(17)重复步骤15;
(18)弃掉离心柱,样本于-80℃冰箱保存。
1.4miRNA文库建立
根据仪器及试剂盒的使用说明,以单个测序样品输入1-500ng总RNA量作为材料以构建本次测序的miRNA文库,使用QIAseq miRNA Library Kit(Qiagen,)生成测序文库,每个测序样品在属性序列中额外加入索引代码,最终经Bioanalyzer 2100(Agilent)和qPCR评估本次小RNA文库构建的质量。按照仪器制造商的指导,使用TruSeq PE-Cluster Kitv3-cBot-HS(Illumina)通过额外加入索引编码进行样本聚类,根据聚类结果,利用Illumina Hiseq平台上对本次建立的小RNA文库测序,并生成成对端reads。
1.5测序数据的处理和分析
通过碱基识别对测序平台得到的高通量测序原始图像变更为原始测序序列(RawReads),最终保存为FASTQ格式的数据文件,序列信息和序列相关质量信息被存储在这些文件中。一些接头序列及低质量序列的存在会对测序结果造成影响,因此需要从原始序列将相关序列剔除以确保后续测序分析的准确程度,得到可用于鉴定和表达分析的高质量序列(即Clean Reads),执行如下序列质量控制方案:(1)将各测序样本中质量值不达标的序列剔去;(2)当测序序列中不能够鉴别碱基N占比≥10%时,去掉该序列;(3)测序序列无3’端接头时,去掉该序列;(4)剪切掉3’接头序列;(5)去掉长度<15nt及>35nt的测序序列;
最终计算Clean Reads的Q30作为质控标准。所有的后续分析都是基于质控方案处理后的高质量序列。Bowtie在遗传物质测序分析中被广泛应用,它能够高效率地将长度较短的序列比对至数据库或是基因组中,通过Bowtie软件,将上述得到的高质量序列(CleanReads)分别与用于获取过滤核糖体RNA(rRNA)的Silva数据库(https://www.arb-silva.de/)、获取转运RNA(tRNA)的GtRNAdb数据库(http://gtrnadb.ucsc.edu/)、获取非编码RNA(no-coding RNA)的Rfam数据库(http://rfam.xfam.org/)和获取重复序列(Repbase)的Repbase数据库(http://www.girinst.org/server/RepBase/index.php)进行序列比对,最终未比对上的序列被被称为未注释序列(Unannotated reads),其中包含了所需miRNA序列信息。
本发明中高活力组和低活力组各三个重复,总共得到原始数据分别为66062578和72832844,去除低质量序列(Low quality)、接头序列(Containing'N'reads)以及长度不在15~35个核苷酸的测序序列,最终用于下一步分析的Clean reads数量分别为47795289和28008495,具体整理结果见表2。表中显示所有样品均无低质量序列,在进行数据过滤后Q30(识别错误率为0.1%)最低为96.96%,表明数据可用于下一步分析。经Bowtie完成序列比对,将sRNA注释到rRNA、tRNA、snRNA、snoRNA、Repbase以及包含miRNA的Unannotatedreads。两组的sRNA分类注释情况见表3。Unannotated reads是后续用于miRNA鉴定与分析的部分,两组中的Unannotated reads分别占过滤后数据的76.37%和64.62%。
表2原始数据过滤和质控
表3sRNA分类注释
1.6miRNA鉴定
将上述得到未注释序列通过Bowtie软件与猪的参考基因(Sus scrofa 11.1)进行比对,以获得测序序列在猪参考基因组中的位置信息(Mapped Reads)。Mapped reads经过与miRBase(v22)数据库中的miRNA成熟序列比对,当Mapped reads和数据库中成熟形态的已知miRNA序列比对吻合即鉴定为已知miRNA。基于miRNA在加工形成中的特点,对没有与miRBase(v22)数据库比对成功的序列通过miRDeep2软件预测未知miRNA。通过QsRNA-seq的方法进行建库,建立测序文库的过程中采用UMI技术以达到降低聚合酶链式反应中的误差,使miRNA的表达量更为精确,TPM算法用于miRNA表达量的归一化处理,公式为:TPM=Readcount/Mapped read*1000000。等式中Read count为鉴定为某一miRNA的序列数量;MappedReads为鉴定为miRNA全部序列数量。
经过Unannotated reads与猪基因组数据比对,测序数据的序列长度分布情况的分析如图2。可以发现两组中鉴定的miRNA大多集中于21~23nt,这一部分miRNA在高活力组和低活力组分别占比66.58%和70.80%。峰值均产生在miRNA典型长度值22nt,在高活力组和低活力组分别占35.73%和38.12%。
通过Unannotated reads与miRBase(v22)比对已知miRNA并利用miRDeep2预测未知miRNA。鉴定到的已知miRNA及预测的未知miRNA在各个样品中情况如表4所示,在各组中的分布情况如图3所示。两组测序数据共鉴定到338个已知miRNA,预测到508个新miRNA。在高活力组中单独表达的包括20个已知miRNA和94个新miRNA。在低活力组中单独表达的包括已知13个已知miRNA和47个未知miRNA。
表4测序样品miRNA鉴定结果
对所有鉴定到的miRNA表达量归一,计算方法使用TMP算法,处理后对每个测序样品的miRNA整体表达模式进行分析,结果如图4。由图可知,两组样品的表达概率峰值均发生TPM值0~10范围内,低活力组三个样品的峰值整体相较于高活力组右移,这表明峰值附近所表示的miRNA在低活力样品中拥有更高的表达量。
1.7miRNA表达分析
使用R语言软件通过edgeR对高活力组和低活力组鉴定到的miRNA进行表达量分析和差异分析,差异miRNA的检测从差异倍数(Fold Change,FC)和显著性p值(p-value)两个方面进行筛选,差异倍数为测序样品组间表达量的比值。显著性p值表示组件表达量无差异的概率。当这两个条件的数值满足FC>1.5、p-value<0.05即为差异表达miRNA。同时为了得到更有生物学功能意义的分析结果,在进行表达分析时剔除低表达量miRNA,最终仅留平均表达量高于1的miRNA用于进一步表达分析。
根据鉴定到的miRNA表达量数据,对高活力组和低活力组的外泌体miRNA进行差异表达分析。以表达变化倍数>1.5且统计学差异P<0.05作为筛选组间差异表达miRNA的标准。经过筛选,共发现49个显著性差异miRNA,与高活力组相比,低活力组中存在38个高表达miRNA,11个低表达miRNA,如图5所示。在49个差异表达miRNA中有17个已知miRNA,32个未知miRNA,根据TPM表达量对已知的差异表达miRNA进行聚类分析,ssc-miR-10a-3p、ssc-miR-139-5p、ssc-miR-148b-5p、ssc-miR-31、ssc-miR-345-3p、ssc-miR-362、ssc-miR-500-5p在高活力组中高表达,ssc-miR-143-3p、ssc-miR-10386、ssc-miR-122-5p、ssc-miR-142-5p、ssc-miR-199a-3p、ssc-miR-199b-3p、ssc-miR-223、ssc-miR-451、ssc-miR-486、ssc-miR-708-5p在低活力组高表达,结果如图6和图7所示。
1.8靶基因预测和通路富集分析
根据鉴定的差异表达miRNa,基于3'UTR作为靶向序列通过TargetScan数据库和miRDB数据库来预测miRNA的靶基因。基因本体(gene ontology,GO)是基因研究中使用常见的数据库,其有助于了解靶基因在细胞组分、生物过程、分子功能中的富集情况,如发生作用的位置、发挥的功能等。京都基因和基因组百科全书(kyoto Encyclopedia of Genesand Genomes,KEGG)是分析基因在通路中富集状况的数据库资源,有助于将基因串联起来形成通路网络进行整体分析。为了进一步了解靶基因之间存在的关系,本发明通过GO和KEGG通路富集分析发掘其在机体中的生物学功能。
依据miRNA碱基互补配对的原则,使用TargetScan和miRDB两种靶基因预测软件对基于3‘UTR为靶向序列的17个已知差异表达miRNA进行靶基因预测,在两组测序样品中共得到靶基因313个。为探究这些基因在体内发挥调节作用的位置和方式,本发明通过GO数据库和KEGG数据库作进一步富集分析,结果如图8所示。
如图8-10所示,GO分析主要包括细胞组分(cellular component,CC)、分子功能(molecular function,MF)、生物过程(biological process,BP)三部分,细胞组分分析结果显示靶基因主要作用位置发生在细胞核、细胞质、细胞质基质中。ATP结合是分子功能中靶基因富集最多的条目,其次是RNA聚合酶Ⅱ与序列特异性DNA的结合、转录激活活性、DNA结合。在生物过程中富集基因最多的依次为RNA聚合酶Ⅱ的转录调控、基因表达正调控、细胞内信号转导,其中富集基因最多的前20位条目中有4项涉及蛋白质磷酸化。
如图11所示,进行KEGG通路富集分析,气泡色相代表富集分析的显著性,气泡体积代表富集分析后在该通路发现的基因数目,结果显示显著性最高的5条通路依次是基于RNA聚合酶Ⅱ的转录活化剂活性、ATP结合、RNA聚合酶Ⅱ与DNA的特异性结合、酪氨酸磷酸酶活性、胰岛素受体结合,其中富集最显著最多的前20条通路中有8项与RNA聚合酶及转录活动相关。
实施例2、引物设计和qPCR反应
本发明选取上述分析结果中的6个差异表达miRNA进行qPCR试验,从而验证miRNA测序分析的准确性,进而筛选出能够用于鉴定公猪精液质量的miRNA标志物。
2.1总RNA提取
该步骤操作与1.3相同。
2.2反转录体系配制
试验使用miRcute增强型miRNA cDNA第一链合成试剂盒(TIANGEN),该试剂盒原理是利用加尾法法来进行cDNA的反转录,反转录体系总体积为20ul,如下表5所示。反应发生条件依次为:42℃温度环境下充分反应60分钟用于在miRNA尾端添加PolyA,并进行逆转录形成cDNA;95℃温度环境下反应3分钟用于PolyA聚合酶和逆转录酶失活。得到的cDNA保存-20℃冰箱。
表5反转录体系
2.3引物设计
从miRBase数据库(https://www.mirbase.org/)获取miRNA序列,将序列中的U替换为T,使用加尾法设计miRNA上游引物,为配合后续试剂盒需保证上游引物的退火温度Tm在65℃左右,上游引物经上海生工加工合成,具体序列见下表6。
表6上游引物设计
2.4qPCR反应体系
试验使用miRcute增强型miRNA荧光定量检测试剂盒(TIANGEN)进行选定miRNA的qPCR反应,通过QuantStudio3型实时荧光定量PCR系统进行定量检测,反应体系配制见下表7所示。反应程序按如下设定:起始模板经95℃高温变性15分钟,1个循环;PCR循环中模板变性:PCR循环中94℃高温模板变性20秒;60℃退火、延伸34秒;后两个部分共40~45个循环。
表7qPCR反应体系
2.5数据处理与分析
以U6作为内源性对照,采用比较Ct法(ΔΔCt)分析每个miRNA在两组间的差异倍数作为相对表达水平的衡量指标。最终将相对表达结果与miRNA-seq数据进行比较。
对1.7中miRNA表达分析结果的准确程度,本发明选取上述分析结果中的6个差异表达miRNA进行qPCR试验,其中包含5个上调表达miRNA,1个下调表达miRNA。检测结果经分析处理后以差异倍数(Fold Change)表示,差异倍数表示低活力公猪精液精浆外泌体中的表达量与高活力公猪精液精浆外泌体中的表达量的比值,然后与miRNA测序结果进行比较,如图9所示。结果显示6个差异表达miRNA与测序结果表达变化是同步的,miRNA测序分析具有准确性。其中5个上调表达的miRNA的差异倍数均>1.5,1个下调表达的miRNA的差异倍数<0.85。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 一种与鸡精子活力和受精率相关的miRNA及其应用
- 一种与公猪精子活力和总精子数相关的分子标记及应用
- 一种与公猪精子活力和总精子数相关的分子标记及应用