稳定同位素2H(D)标记25-羟基维生素D2的合成方法
文献发布时间:2023-06-19 19:28:50
技术领域
本发明属于有机合成领域,具体涉及一种稳定同位素2H(D)标记25-羟基维生素D2的合成方法。
背景技术
维生素D是体内重要的营养素,维生素D的过量或缺乏都会导致健康问题的发生,其本身不具备生理功能,只有转变为活性形式才能发挥生理活性。维生素D的主要活性形式有:25(OH)D2/D3、1,25-(OH)
目前,检测维生素D的方法有免疫法、放射免疫法、液相色谱法以及液相色谱-串联质谱联用法(LC-MS/MS)等。美国内分泌学会推荐采用25(OH)D水平评价患者体内的维生素D状况,随着LC-MS/MS分析灵敏度的提高,已经可以同时测量25(OH)D2和25(OH)D3,由于检测更加准确、稳定,被认为是检测维生素D的金标准。
在质谱分析中,同位素内标法是以稳定同位素标记化合物作为内标,利用待测物与内标的响应丰度比值(峰面积比)进行线性回归,通过对同位素丰度的准确质谱测量和对加入的内标准确称量,做到对待测物的定量测定。
但是,稳定同位素标记25-羟基维生素D2来源十分有限,目前关于合成稳定同位素标记的25(OH)D2化合物的报道很少,国外有研究人员通过硼酸酯与烯醇三氟甲烷磺酸 Pd( 0) 催化偶联的汇聚法合成了九重氢标记的类似物 27,27,27,26,26,26,28,28,28-d
专利文件CN110272367A公开了一种标记的维生素D2内标化合物的制备方法,以γ-酰胺磷盐等为原料,合成了可用于串联质谱法检测人血清中VD含量标记的维生素D2内标化合物,虽收率可达80%左右,但其原料极难获取。
总的来说,目前合成稳定同位素标记的25-羟基维生素D2主要面临的问题在于:氘代试剂价格昂贵、合成路线长、制备生产难度高。
发明内容
有鉴于此,本发明的目的在于提供一种稳定同位素2H(D)标记的25-羟基维生素D2及其合成方法,丰富了稳定同位素标记25-羟基维生素D2种类,且原料便宜易得,合成路线较短。
本发明的目的是通过以下技术方案来实现的:
一种稳定同位素2H(D)标记的25-羟基维生素D2的合成方法,所述25-羟基维生素D2的结构式如(Ⅰ)所示:
;
所述合成方法包括以下步骤:
S1. 将丙二酸二乙酯与氘代碘甲烷进行加成反应,得到含氘代甲基的中间体(Ⅱ);
S2. 将所述中间体(Ⅱ)还原得到中间体(Ⅲ);
S3. 将所述中间体(Ⅲ)与苯并噻唑-2-3-硫酮进行Mitsunobu反应,得到砜中间体(Ⅳ);
S4. 将所述砜中间体(Ⅳ)进行两次羟基氧化反应和两次甲基锂加成反应,得到中间体(Ⅴ);
/>
S5. 将所述中间体(Ⅴ)进行氧化反应,并对氧化产物进行醇羟基保护,得到亚砜中间体(Ⅵ);
S6. 将所述亚砜中间体(Ⅵ)与
S7. 将所述中间体(Ⅶ)进行四丁基氟化铵(TBAF)催化脱出保护基,催化产物再经氧化反应得到中间体(Ⅷ);
S8. 对所述中间体(Ⅷ)进行叔醇羟基保护,得到中间体(Ⅸ);
S9. 将所述中间体(Ⅸ)与
。
进一步的,步骤S1中,所述加成反应在溶剂中进行,反应温度为室温,反应的pH值为13~15,反应时间为5~7小时;
和/或,步骤S1中,所述溶剂选自乙醇或甲醇中的一种;
和/或,步骤S1中,所述反应的pH值采用甲醇钠或乙醇钠中的一种进行调节;
和/或,步骤S1中,所述丙二酸二乙酯和所述氘代碘甲烷的摩尔比为1:0.8~1.2。
进一步的,步骤S2中,所述还原在溶剂中进行,反应温度为0~10℃,反应时间为5~7小时;
和/或,步骤S2中,所述溶剂选自四氢呋喃(THF)或乙醚中的一种;
和/或,步骤S2中,还原剂选自氢化铝锂(LiAlH
和/或,步骤S2中,所述中间体(Ⅱ)与所述还原剂的摩尔比为1:2~3。
进一步的,步骤S3中,所述Mitsunobu反应在溶剂中进行,
反应温度为室温,反应的时间为4~8小时;
和/或,步骤S3中,所述溶剂选自四氢呋喃或乙醚中的一种;
和/或,步骤S3中,所述中间体(Ⅲ)与苯并噻唑-2-3-硫酮的摩尔比为1:0.8~1。
进一步的,步骤S4中,所述羟基氧化反应和所述甲基锂加成反应均在溶剂中进行,先进行第一次所述羟基氧化反应,氧化产物进行第一次甲基锂加成反应,加成产物再进行第二次所述羟基氧化反应,得到的产物再进行第二次所述甲基锂加成反应;
和/或,步骤S4中,两次所述羟基氧化反应均在室温下进行,反应的时间均为2~4小时,两次所述甲基锂加成反应的反应温度为均为-40℃~0℃,反应时间均为3~5小时;
和/或,步骤S4中,所述羟基氧化反应的溶剂选自二氯甲烷(DCM),二氯乙烷(DCE)或四氢呋喃(THF)中的一种,氧化剂选自Dess-Martin或PCC的一种;
和/或,步骤S4中,所述甲基锂加成反应使用的溶剂选自乙醚(Et
和/或,步骤S4中,进行两次所述羟基氧化反应时,反应物和所述氧化剂的摩尔比均为1:1.3~1.6;
和/或,步骤S4中,进行两次所述甲基锂加成反应时,反应物与甲基锂的摩尔比均为1:2.5~3。
进一步的,步骤S5中,所述氧化反应和所述醇羟基保护均在溶剂中进行,所述氧化反应的反应温度为室温,反应时间为3~5小时,所述醇羟基保护的反应温度为0~10℃,反应的pH值为13~15,反应时间为2~4小时;
和/或,步骤S5中,所述溶剂选自二氯甲烷,二氯乙烷或四氢呋喃中的一种;
和/或,步骤S5中,所述氧化反应的氧化剂选自间氯过氧苯甲酸(mCPBA)或过氧化氢(H
和/或,步骤S5中,所述醇羟基保护的保护试剂选自三甲基氯硅烷,三乙基氯硅烷或叔丁基二甲基氯硅烷中的一种;
和/或,步骤S5中,所述醇羟基保护的pH值采用咪唑进行调节;
和/或,步骤S5中,所述中间体(Ⅴ)与所述氧化剂和所述保护试剂的摩尔比为1:2.5~3:1.5~2.5。
进一步的,步骤S6中,所述反应在溶剂中进行,反应的温度为-78~-58℃,反应的pH值为13~15,反应时间为2~4小时;
和/或,步骤S6中,所述反应使用的溶剂选自四氢呋喃或乙醚中的一种;
和/或,步骤S6中,所述反应的pH值使用六甲基二硅基胺基锂(LiHMDS)或二异丙基氨基锂(LDA)中的一种进行调节;
和/或,步骤S6中,所述亚砜中间体(Ⅵ)与所述
进一步的,所述
进一步的,步骤S7中,所述四丁基氟化铵催化和所述氧化反应均在溶剂中进行,反应的温度均为室温,所述四丁基氟化铵催化的时间为5~7小时,所述氧化反应的时间为1~3小时;
和/或,步骤S7中,所述四丁基氟化铵催化的溶剂选自四氢呋喃和乙醚中的一种,所述中间体(Ⅶ)与所述四丁基氟化铵的摩尔比为1:3~5;
和/或,步骤S7中,所述氧化反应的溶剂选自二氯甲烷和四氢呋喃中的一种,氧化剂选自Dess-Martin和PCC 中的一种,所述催化产物与所述氧化剂的摩尔比为1:1.2~1.6。
进一步的,步骤S8中,所述叔醇羟基保护在溶剂中进行,反应温度为室温,反应的pH值为13~15,反应时间为2~4小时;
和/或,步骤S8中,所述溶剂选自二氯甲烷,二氯乙烷或四氢呋喃中的一种;
和/或,步骤S8中,所述反应的pH值使用咪唑进行调节;
和/或,步骤S8中,保护试剂选自三甲基氯硅烷,三乙基氯硅烷或叔丁基二甲基氯硅烷的一种;
和/或,步骤S8中,所述中间体(Ⅷ)与所述保护试剂的摩尔比为1:1.5~2.5。
进一步的,步骤S9中,所述反应和所述四丁基氟化铵催化脱保护均在溶剂中进行,所述反应的反应温度为-78~-58℃,所述反应的pH值为13~15,反应时间为1~2小时,所述四丁基氟化铵催化脱保护的反应温度为室温,反应时间为4-8小时;
和/或,步骤S9中,所述反应使用的溶剂选自四氢呋喃和乙醚中的一种;
和/或,步骤S9中,所述反应的pH值使用正丁基锂,二异丙基氨基锂和六甲基二硅基胺基锂中的一种进行调节;
和/或,步骤S9中,所述中间体(Ⅸ)与所述
和/或,步骤S9中,所述四丁基氟化铵催化脱保护使用的溶剂选自四氢呋喃和乙醚中的一种,所述产物(Ⅹ)与所述四丁基氟化铵的摩尔比为1:3~5。
进一步的,所述
本发明的有益效果是:
1. 本发明合成条件较为温和,合成路线短,收率较高;
2. 本发明使用的原料便宜易得,较现有技术相比,大幅降低了成本;
3. 本发明合成的稳定同位素2H(D)标记的25-羟基维生素D2,氘原子取代基位于烷基侧链的中间位置,与部分市售产品氘原子取代基在烷基末端相比,有效避免了其易被氧化代谢的缺点。
附图说明
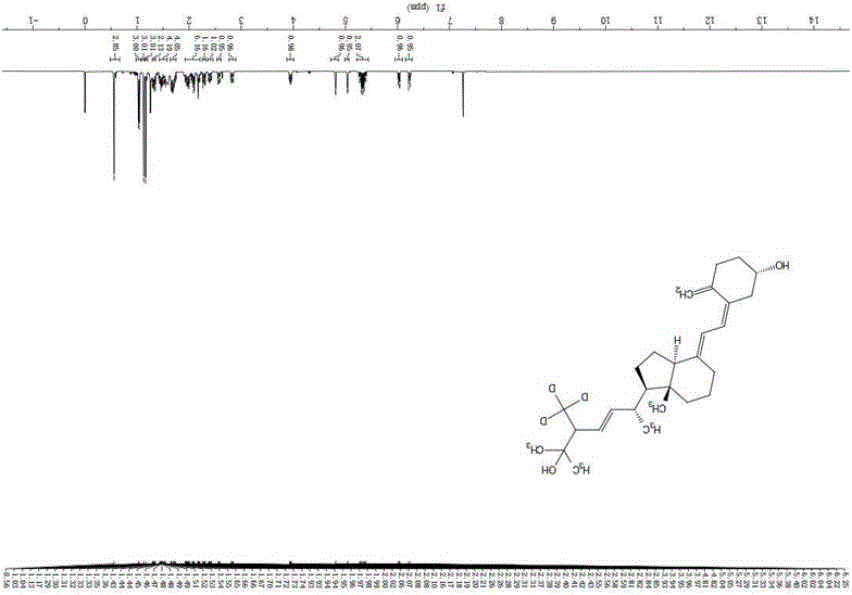
图1 :砜中间体(Ⅳ)的核磁共振氢谱图;
图2:实施例4中间产物4-苯并噻唑-2-硫基-3-甲基丁烷-2-醇的核磁共振氢谱图;
图3:实施例4中间产物4-苯并噻唑-2-硫基-3-甲基丁烷-2-酮的核磁共振氢谱图;
图4:实施例7中催化产物的核磁共振氢谱图;
图5:中间体(Ⅷ)的核磁共振氢谱图;
图6:中间体(Ⅸ)的核磁共振氢谱图;
图7:产物(Ⅹ)的核磁共振氢谱图;
图8:产品(Ⅰ)的核磁共振氢谱图;
图9:产品(Ⅰ)的核磁共振碳谱图;
图10:产品(Ⅰ)的高效液相色谱-质谱联用分析图谱;
图11:产品(Ⅰ)的液相色谱串联质谱法(LC-MS/MS)分析图谱;
图12:谱育公司的VD2-d3标准品的液相色谱串联质谱法(LC-MS/MS)分析图谱。
具体实施方式
下面结合附图进一步详细描述本发明的技术方案,但本发明的保护范围不局限于以下所述。
实施例1
取60.6mmol丙二酸二乙酯至乙醇中,加入60.6mmol氘代碘甲烷,使用甲醇钠调节pH值至14,在室温下反应6小时,反应结束后减压旋干溶剂,同时加水(100 mL),使用乙酸乙酯萃取(150 mL× 3),萃取液经无水硫酸钠干燥后,过滤,旋干溶剂,采用硅胶柱层析分离纯化(PE : EA = 50 : 1),得到为无色透明液体的中间体(Ⅱ),即2-甲基丙二酸二乙酯8.2g,产率为76.6%。
实施例2
将氢化铝锂(1.9 g,50.8 mmol,2.5eq)溶于干燥的THF(90 mL)中,在氮气保护下,降温至0℃,然后取实施例1制得的产物2-甲基丙二酸二乙酯(3.6 g,20.3 mmol,1.0eq)的THF溶液(15 mL)滴加入其中,滴加完毕后,于此温度下反应6小时,薄层层析硅胶板(TLC)显示(PE : EA = 10 : 1)原料反应完全。缓慢滴加水淬灭反应,加入无水硫酸钠干燥,过滤,减压旋干溶剂,得到无色油状物中间体(Ⅲ),即氘代2-甲基丙烷-1,3-二醇粗品1.5 g,粗品未经进一步纯化直接投下一步反应。
实施例3
取实施例2制得的产物氘代2-甲基丙烷-1,3-二醇粗品11.8 mmol,与苯并噻唑-2-3-硫酮10.7 mmol,三苯基膦11.8mmol,DIAD11.8mmol溶于100mL干燥的THF中,室温条件下反应过夜,TLC显示(PE : EA = 5 : 1)原料反应完全,加水(80 mL),采用乙酸乙酯萃取(100 mL × 3),经无水硫酸钠干燥,过滤,旋干溶剂,硅胶柱层析分离纯化(PE : EA = 15 :1),得到黄色油状物2.1 g(产率80.7%),产物为氘代3-苯并噻唑-2-硫基-2-甲基丙醇,即砜中间体(Ⅳ)。图1为砜中间体(Ⅳ)的核磁共振氢谱图,分析如下:1H NMR (400 MHz,Chloroform-d) δ 7.84 (dt, J = 8.3, 0.9 Hz, 1H), 7.76 – 7.70 (m, 1H), 7.42(tt, J = 8.2, 1.1 Hz, 1H), 7.35 – 7.28 (m, 1H), 4.10 (s, 1H), 3.63 (ddd, J =14.6, 9.5, 4.1 Hz, 2H), 3.45 (ddd, J = 18.6, 13.0, 6.8 Hz, 2H), 2.17 (q, J =6.2, 5.6 Hz,1H)。
实施例4
取实施例3制得的产物氘代3-苯并噻唑-2-硫基-2-甲基丙醇2.9 mmol至溶剂二氯甲烷中,加入4.4 mmol Dess-Martin氧化剂,室温条件下反应4小时,加水30 mL,使用二氯甲烷萃取(50 mL × 3),经无水硫酸钠干燥,过滤,旋干溶剂,得到黄色固体(0.70 g)氘代3-苯并噻唑-2-硫基-2-甲基丙醛粗品,粗品未经进一步纯化直接投下一步反应。
取上述制得的氘代3-苯并噻唑-2-硫基-2-甲基丙醛的粗品2.9 mmol至35 mL绝对乙醚中,加入7.3 mmol金属试剂甲基锂,设置反应温度为-40℃,反应时间为4小时。加入饱和氯化铵水溶液淬灭反应,加水(20 mL),乙酸乙酯萃取(30 mL × 3),无水硫酸钠干燥,过滤,旋干溶剂,硅胶柱层析分离纯化(PE :EA = 10 : 1),得到黄色油状物4-苯并噻唑-2-硫基-3-甲基丁烷-2-醇(563 mg),产率75.5%。图2为该产物的核磁共振氢谱图,分析如下:1HNMR (400 MHz, Chloroform-d) δ 7.84 (ddd, J =8.2, 4.5, 1.1 Hz, 1H), 7.76 –7.69 (m, 1H), 7.41 (dd, J = 8.3, 7.1 Hz, 1H), 7.33 – 7.27(m, 1H), 4.15 (br,1H), 4.07 (dd, J = 14.2, 4.6 Hz, 0.3H), 3.89 (dd, J = 14.2, 4.6 Hz, 0.7H),3.75 (dq, J = 8.7, 6.3 Hz, 0.3H), 3.61 (dq, J = 8.7, 6.3 Hz,0.7H), 3.33 (dd,J = 14.2, 3.5 Hz, 0.7H), 3.05 (dd, J = 14.2, 3.5 Hz, 0.3H), 1.96 (dt, J =8.6, 4.0 Hz, 0.7H), 1.83 (dt, J = 8.6, 4.0 Hz, 0.3H), 1.22 (d, J= 6.2 Hz,2.1H), 1.19 (d, J = 6.2 Hz, 0.9H).
再取制得的4-苯并噻唑-2-硫基-3-甲基丁烷-2-醇9.0 mmol溶于二氯甲烷中,加入13.4 mmol Dess-Martin氧化剂,设置反应温度为室温,反应时间为2小时,加水(80 mL),二氯甲烷萃取(100 mL × 3),无水硫酸钠干燥,过滤,旋干溶剂,硅胶柱层析分离纯化(PE :EA = 10 : 1),得到黄色油状物4-苯并噻唑-2-硫基-3-甲基丁烷-2-酮(2.2 g),产率95.5%。图3为该产物的核磁共振氢谱图,分析如下:1H NMR (400 MHz, Chloroform-d) δ7.90 – 7.82 (m, 1H), 7.75 (dd, J = 8.0, 1.2 Hz, 1H), 7.46 – 7.37 (m, 1H),7.30 (td, J = 7.7, 7.2, 1.2 Hz, 1H), 3.62(dd, J = 13.5, 7.2 Hz, 1H), 3.37(dd, J = 13.5, 6.3 Hz, 1H), 3.12 (t, J = 6.8 Hz, 1H), 2.24 (s, 3H).
取制得的4-苯并噻唑-2-硫基-3-甲基丁烷-2-酮8.7 mmol至100 mL绝对乙醚中,加入21.8 mmol金属试剂甲基锂,设置反应温度为-40℃,反应时间为4小时,加入饱和氯化铵水溶液淬灭反应,加水(100 mL),乙酸乙酯萃取(120 mL × 3),无水硫酸钠干燥,过滤,旋干溶剂,硅胶柱层析分离纯化(PE : EA = 6 : 1),得到黄色固体4-苯并噻唑-2-硫基-2,3-二甲基丁烷-2-醇,即中间体(Ⅴ)(2.1 g),产率91.3%。
实施例5
取实施例4制得的4-苯并噻唑-2-硫基-2,3-二甲基丁烷-2-醇4.5mmol至100mL二氯甲烷中,加入13.5mmolmCPBA,室温反应4 h,TLC显示(PE : EA = 4 : 1)原料基本反应完全。加入饱和硫代硫酸钠水溶液淬灭反应,加水(50 mL),二氯甲烷萃取(60 mL × 3),有机层分别用饱和碳酸氢钠、水和饱和食盐水洗涤(80 mL × 1),无水硫酸钠干燥,过滤,旋干溶剂,硅胶柱层析分离纯化(PE : EA = 5 : 1),得到中间体无色透明油状物(1.1 g)。将此中间体溶于溶于二氯甲烷(60 mL)中,降温至0℃,加入咪唑(490 mg,7.2 mmol,2.0eq)和三甲基氯硅烷(0.95 mL,7.2 mmol,2eq),于此温度下搅拌反应3 h,TLC显示(PE : EA = 5 :1)原料反应完全,加水(50 mL),二氯甲烷萃取(60 mL × 3),无水硫酸钠干燥,过滤,旋干溶剂,硅胶柱层析分离纯化(PE :EA = 8 : 1),得到乳白色固体产物2-(2,3-二甲基-3-(三甲基硅氧基)丁基磺酰基)苯并噻唑即亚砜中间体(Ⅵ)(1.3 g),产率96.3%。
实施例6
取实施例5制得的2-(2,3-二甲基-3-(三甲基硅氧基)丁基磺酰基)苯并噻唑3.12mmol至溶剂50 mL四氢呋喃中,降温至-78℃加入4.8 mmol六甲基二硅基胺基锂调节pH值至14,搅拌30 min后,滴加化合物(S)-2-((1R,3aR,4S,7aR)-7a-methyl-4-((triethylsilyl)oxy)octahydro-1H-inden-1-yl)propanal3.85 mmol的THF溶液(15 mL),
完毕于此温度搅拌2 h,TLC显示(PE : EA = 5 : 1)原料反应完全,加入饱和氯化铵水溶液淬灭反应,加水(50 mL),乙酸乙酯萃取(80 mL × 3),无水硫酸钠干燥,过滤,旋干溶剂,得到产物(((6R,Z)-2,3-dimethyl-6-((1R,3aR,4S,7aR)-7a-methyl-4-((triethylsilyl)oxy)octahydro-1H-inden-1-yl)hept-4-en-2-yl)oxy)trimethylsilane为无色透明液体,粗品(1.56 g),即中间体(Ⅶ),粗品未经进一步纯化直接投下一步反应。
实施例7
取实施例6制备的中间体(Ⅶ)粗品2.8 mmol溶于THF(50 mL)中,加入11.3mmolTBAF,在室温条件下反应时间6h,TLC显示(PE : EA = 5 : 1)原料反应完全,加水(50mL),乙酸乙酯萃取(80 mL× 3),无水硫酸钠干燥,过滤,旋干溶剂,硅胶柱层析分离纯化(PE : EA = 10 : 1),得到催化产物(1R,3aR,4S,7aR)-1-((2R,Z)-6-hydroxy-5,6-dimethylhept-3-en-2-yl)-7a-methyloctahydro-1H-inden-4-ol为乳白色固体(0.39 g),产率47.0%。图4为该产物的核磁共振氢谱图,分析如下:1HNMR (400 MHz, Chloroform-d)δ 5.40 – 5.25 (m, 2H), 4.08 (q, J = 2.8 Hz, 1H), 2.13 – 2.03 (m, 2H), 2.00 –1.95 (m, 1H), 1.86 – 1.76 (m, 2H), 1.73 – 1.63 (m, 1H), 1.57 – 1.51(m, 1H),1.47 – 1.20 (m, 6H), 1.17 (s, 3H), 1.13 (s, 3H), 1.01 (d, J = 6.6 Hz, 3H),0.95 (s, 3H), 0.90 – 0.81 (m, 1H).
取上述产物1.3 mmol溶于于溶剂二氯甲烷中加入2.0 mmol Dess-Martin试剂,于室温反应2 h,TLC显示(PE : EA = 3 : 1)原料反应完全。加水(10 mL),二氯甲烷萃取(20mL× 3),无水硫酸钠干燥,过滤,旋干溶剂,硅胶柱层析分离纯化(PE : EA = 5 : 1),得到乳白色固体(1R,3aR,7aR)-1-((2R,Z)-6-hydroxy-5,6-dimethylhept-3-en-2-yl)-7a-methylhexahydro-1H-inden-4(2H)-one,即中间体(Ⅷ)(0.35 g),产率92.8%。图5为中间体(Ⅷ)的核磁共振氢谱图,分析如下:1HNMR (400 MHz, Chloroform-d) δ 5.40 – 5.25 (m,2H), 2.51 – 2.23 (m, 3H), 2.17 – 1.84 (m, 5H), 1.78 – 1.69 (m, 3H), 1.56 –1.26(m, 3H), 1.16 (d, J = 3.7 Hz, 3H), 1.13 (d, J = 3.3 Hz, 3H), 1.07 (d, J =6.6 Hz, 1.5H), 1.05 (s, 1.5H), 1.01 (d, J = 6.6 Hz, 1.5H), 0.65 (s, 1.5H).
实施例8
取实施例7制得的产物中间体(Ⅷ)1.1 mmol于溶剂二氯甲烷(10 mL)中,加入2.2mmol三甲基氯硅烷,加入2.2 mmol咪唑调节pH值至14,在室温条件下搅拌反应3 h,TLC显示(PE : EA = 3 : 1)原料反应完全,加水(10 mL),二氯甲烷萃取(10 mL × 3),无水硫酸钠干燥,过滤,旋干溶剂,硅胶柱层析分离纯化(PE :EA = 5 : 1),得到氘代(1R,3aR,7aR)-1-((2R,Z)-5,6-dimethyl-6-((trimethylsilyl)oxy)hept-3-en-2-yl)-7a-methylhexahydro-1H-inden-4(2H)-one为无色透明液体,即中间体(Ⅸ)(0.37 g),产率92.5%。图6为中间体(Ⅸ)的核磁共振氢谱图,分析如下:1H NMR (400 MHz, Chloroform-d)δ 5.28 – 5.03 (m, 2H), 2.35 (dd, J = 11.2, 7.6 Hz, 1H), 2.22 – 2.08 (m, 2H),2.02 – 1.79(m, 4H), 1.67 – 1.56 (m, 2H), 1.53 – 1.15 (m, 5H), 1.09 – 0.97 (m,6H), 0.97 – 0.93 (m, 3H), 0.99 – 0.87 (m, 0.7H), 0.56 – 0.55(m, 2.3H), 0.06 –-0.02 (m, 9H).
实施例9
取0.43 mmol (S,Z)-(2-(5-((tert-butyldimethylsilyl)oxy)-2-methylenecyclohexylidene)ethyl)diphenylphosphineoxide(该化合物已经商品化)溶于干燥的THF(10 mL)中,降温至-78℃,加入正丁基锂(0.41 mL,0.65 mmol,1.6 M,1.5eq),搅拌1 h后,滴加实施例8制得的即中间体(Ⅸ)0.43 mmol的10 mL四氢呋喃溶液,完毕于此温度搅拌2 h,TLC显示(PE : EA = 5 : 1)原料反应完全,加入饱和氯化铵水溶液淬灭反应,加水(10 mL),乙酸乙酯萃取(30 mL × 3),无水硫酸钠干燥,过滤,旋干溶剂,硅胶柱层析分离纯化(PE),得到氘代产物tert-butyl(((1S,Z)-3-((E)-2-((1R,3aS,7aR)-1-((2R,Z)-5,6-dimethyl-6-((trimethylsilyl)oxy)hept-3-en-2-yl)-7a-methylhexahydro-1H-inden-4(2H)-ylidene)ethylidene)-4-methylenecyclohexyl)oxy)dimethylsilane为乳白色固体(132 mg),即产物(Ⅹ),产率51.5%。图7为产物(Ⅹ)的核磁共振氢谱图,分析如下:1HNMR (400 MHz, Chloroform-d) δ 6.06 (d, J = 11.2 Hz, 1H), 5.91 (d, J = 11.2Hz, 1H), 5.22 – 5.06(m, 2H), 4.90 (t, J = 1.9 Hz, 1H), 4.67 (dd, J = 2.7, 1.3Hz, 1H), 3.72 (tt, J = 8.8, 4.0 Hz, 1H), 2.73 (dd, J = 11.7, 3.8 Hz, 1H),2.39 – 2.21 (m, 2H), 2.13 (dd, J = 12.9, 9.0 Hz, 1H), 2.03 – 1.77 (m, 6H),1.60 – 1.54(m, 2H), 1.48 – 1.33 (m, 4H), 1.25 – 1.12 (m, 4H), 1.06 (s, 3H),1.01 (s, 3H), 0.91 (d, J = 6.6 Hz, 3H), 0.79 (s, 9H), 0.46 (s, 3H), 0.00 –-0.10 (m, 15H).
取产物(Ⅹ)0.17 mmol至5 mL四氢呋喃中,加入0.68 mmolTBAF,于室温搅拌反应6h,TLC显示(PE : EA = 3 : 1)原料反应完全,加水(10 mL),乙酸乙酯萃取(20 mL × 3),无水硫酸钠干燥,过滤,减压旋干溶剂,硅胶柱层析分离纯化(PE :EA = 5 : 1),得到氘代(1S,Z)-3-((E)-2-((1R,3aS,7aR)-1-((2R,Z)-6-hydroxy-5,6-dimethylhept-3-en-2-yl)-7a-methylhexahydro-1H-inden-4(2H)-ylidene)ethylidene)-4-methylenecyclohexanol,即产品(Ⅰ):稳定同位素2H(D)标记25-羟基维生素D2,为乳白色固体(50 mg),产率72.8%,总产率为。图8为产品(Ⅰ)的核磁共振氢谱图,分析如下:1H NMR(400 MHz, Chloroform-d) δ 6.23 (d, J =11.2 Hz, 1H, 11-H), 6.03 (dd, J = 11.3,1.9 Hz, 17-H), 5.33 (qd, J = 15.3, 7.9 Hz, 2H, 15, 16-H), 5.04 (d, J = 2.7Hz, 1H, 24-H), 4.81 (d, J = 2.5 Hz, 1H,24-H), 3.95 (tt, J = 7.4, 3.6 Hz, 1H,20-H), 2.83 (dd, J = 12.0, 4.2 Hz, 1H, 26-H), 2.57 (dd, J = 13.1, 3.8 Hz,1H), 2.40 (ddd, J = 13.1, 7.9, 4.7 Hz, 1H),2.32 – 2.26 (m, 1H), 2.25 – 1.87(m, 6H), 1.75 – 1.64 (m, 4H), 1.57 – 1.43 (m, 4H), 1.36 – 1.29 (m, 2H), 1.17(s, 3H, 28-H), 1.13 (s, 3H, 30-H),1.04 (d, J = 6.6 Hz, 3H, 14-H), 0.56 (s,3H, 10-H).
图9为产品(Ⅰ)的核磁共振碳谱图,分析如下:
对产品(Ⅰ)进行高效液相色谱-质谱联用分析,图10为分析图谱,质谱中C-OH键裂分,得到的精确分子量(Exact Mass)为398.3502,做出来的质谱分子离子峰为398.35001,确认为所需化合物。
使用甲醇作为稀释液,分别配置浓度为1ng/mL的本发明合成的2H(D)标记的25-羟基维生素D2标准品和购买自谱育公司的VD2-d3标准品,对二者进行液相色谱串联质谱法(LC-MS/MS)分析,测试其作为内标化合物的方法检出限,图11为本发明产品(Ⅰ)的分析图谱,图12为谱育公司的VD2-d3标准品的分析图谱,结果显示二者信噪比分别为17(p-p)和19(p-p),相差不大且均大于10,说明以本发明产品作为内标化合物,灵敏度满足定量要求。
以上所述仅是本发明的优选实施方式,应当理解本发明并非局限于本文所披露的形式,不应看作是对其他实施例的排除,而可用于各种其他组合、修改和环境,并能够在本文所述构想范围内,通过上述教导或相关领域的技术或知识进行改动。而本领域人员所进行的改动和变化不脱离本发明的精神和范围,则都应在本发明所附权利要求的保护范围内。
- 同时检测人末梢血中25-羟基维生素D3和25-羟基维生素D2含量的方法
- 同时检测人末梢血中25-羟基维生素D3和25-羟基维生素D2含量的方法