DNA甲基转移酶结合胞嘧啶脱氨酶介导的DNA中5-甲基胞嘧啶的单碱基定位分析方法
文献发布时间:2023-06-19 19:28:50
技术领域
本发明属于生物技术领域,涉及一种DNA甲基转移酶结合胞嘧啶脱氨酶介导的DNA中5-甲基胞嘧啶的单碱基定位分析方法和试剂盒。
背景技术
5-甲基胞嘧啶(5mC)是哺乳动物中最普遍的表观遗传学修饰。5mC在调控基因表达方面发挥着重要的作用。局部或整体水平下DNA甲基化图谱的改变与多种生物进程和病理过程有关,如胚胎发育、X染色体失活和肿瘤发生。在哺乳动物的基因组中,5mC主要存在于CG二核苷酸序列中,由DNA甲基转移酶(DNMTs)利用S-腺苷-L-甲硫氨酸(SAM)作为甲基供体催化形成。
在哺乳动物基因组中,大约70-80%的CG二核苷酸中的胞嘧啶是甲基化修饰的,准确检测5mC对疾病的诊断有着十分深远的意义。目前,5mC的检测分为两类,包括5mC的整体含量检测和测序分析。已有的检测DNA中5mC整体含量的方法主要包括液相色谱/质谱法(LC/MS)、薄层色谱法(TLC)和电化学检测法。这些方法一般都需要对DNA进行水解消化,不能提供5mC在DNA序列中的位置信息。5mC的测序方法中,基于免疫沉淀的测序方法已被用于DNA中5mC的定位分析,如甲基化DNA免疫沉淀测序(MeDIP-seq)和甲基CpG结合域测序(MBD-seq)等。然而,这些基于免疫亲和富集的方法无法以单碱基分辨率检测5mC,且更偏向于富集高甲基化区域。基于限制性内切酶的方法(如MRE-seq和Methyl-MAPS)能够对5mC进行单碱基分辨率的分析,但其对序列特异性内切酶的依赖限制了CG的覆盖率,并且不完全酶切可能导致高假阳性结果。
亚硫酸氢盐测序(BS-seq)目前已被广泛用于5mC的定位分析,在BS-seq中,胞嘧啶在亚硫酸氢盐处理后被脱氨为尿嘧啶,但5mC抵抗脱氨,不会发生转化。通过测序可以区分C和5mC,因为它们分别被读作胸腺嘧啶和胞嘧啶。然而,BS-seq需要苛刻的化学脱氨条件,从而导致高达99.9%的DNA被降解。随后,有人开发了TET辅助的吡啶硼烷测序(TAPS)法用于5mC的测序。在TAPS中,5mC被TET蛋白氧化为5-羧基胞嘧啶(5caC),然后5caC被吡啶硼烷还原为二氢尿嘧啶。二氢尿嘧啶在随后的PCR扩增中与腺嘌呤配对,因此实现5mC到胸腺嘧啶的转变,从而实现5mC的测序。最近有人开发了一种EM-seq的方法来绘制5mC。在EM-seq中,人载脂蛋白B mRNA编辑催化亚基3A(APOBEC3A,A3A)脱氨酶被用来将胞嘧啶脱氨形成尿嘧啶;而5mC被TET蛋白氧化成5caC,转化后的5caC对A3A脱氨有抵抗作用。这种技术依赖于DNA的酶促脱氨,因此不需要进行亚硫酸氢盐处理。然而,EM-seq涉及三种酶和两个不同的测序库的制备,方法复杂。此外,TAPS测序法和EM-seq测序法均涉及TET蛋白氧化5mC为5caC,然而,TET蛋白不能完全将5-mC氧化为5caC,导致假阴性结果的产生。
因此,如何一种有必要开发一种无需亚硫酸氢盐处理、单碱基分辨率和定量检测5mC的方法。
发明内容
为了解决所述技术问题,本发明提供了一种DNA甲基转移酶结合胞嘧啶脱氨酶介导的DNA中5-甲基胞嘧啶的单碱基定位分析方法,不依赖于亚硫酸氢盐处理,且具有高选择性,操作简单。
为了实现上述目的,本发明采用如下技术方案:
在本发明的第一方面,提供了一种DNA甲基转移酶结合胞嘧啶脱氨酶介导的DNA中5-甲基胞嘧啶的单碱基定位分析方法,所述方法包括:
采用DNA甲基转移酶对待检DNA进行羧甲基化标记反应,后变性处理以使DNA甲基转移酶灭活而终止反应,获得羧甲基化标记的DNA;
采用胞嘧啶脱氨酶对所述羧甲基化标记DNA进行脱氨基反应,后进行高温灭活处理以使胞嘧啶脱氨酶灭活而终止反应,获得脱氨基DNA样品;
将所述脱氨基DNA样品进行PCR反应,获得扩增产物;
将所述扩增产物测序,获得5-甲基胞嘧啶的位点信息。
进一步地,所述羧甲基化标记反应的条件为:双链DNA在含Tris-HCl 5~20mM、pH7.0~8.5、NaCl 20~100mM、二硫苏糖醇0.2~2mM、EDTA 0.2~2mM、羧基-S-腺苷-L-甲硫氨酸80~640mM、M.MpeI-N374K蛋白1~10μM的反应体系中,于30~45℃反应0.5~6小时。
进一步地,所述变性处理的方法为:在0~60% DMSO存在下,将DNA在90~98℃高温下孵育2~30分钟。
进一步地,所述脱氨基反应的条件为:DNA在含2-(N-吗啉)乙磺酸10~30mM、Triton X-100 0.05~0.5%、pH 5.5~7.5、A3A蛋白1~10μM的反应体系中,于30~45℃反应0.5~4小时。
进一步地,所述高温灭活处理的条件包括:于92~98℃水浴中孵育5~20min。
在本发明的第二方面,提供了一种DNA甲基转移酶和胞嘧啶脱氨酶的组合在对DNA中5-甲基胞嘧啶进行单碱基分辨率定位的分析方法中的应用。
在本发明的第三方面,提供了一种用于对DNA中5-甲基胞嘧啶进行单碱基分辨率定位的试剂盒,所述试剂盒包括DNA甲基转移酶和胞嘧啶脱氨酶。
进一步地,所述试剂盒还包括:
采用DNA甲基转移酶进行羧甲基化的反应体系:Tris-HCl 5~20mM、pH 7.0~8.5、NaCl 20~100mM、二硫苏糖醇0.2~2mM、EDTA0.2~2mM、羧基-S-腺苷-L-甲硫氨酸80~640mM、M.MpeI-N374K蛋白1~10μM;
采用胞嘧啶脱氨酶进行脱氨基的反应体系:DNA在含2-(N-吗啉)乙磺酸10~30mM、Triton X-100 0.05~0.5%、pH 5.5~7.5、A3A蛋白1~10μM的反应体系中,于30~45℃反应0.5~4小时。
本发明实施例中的一个或多个技术方案,至少具有如下技术效果或优点:
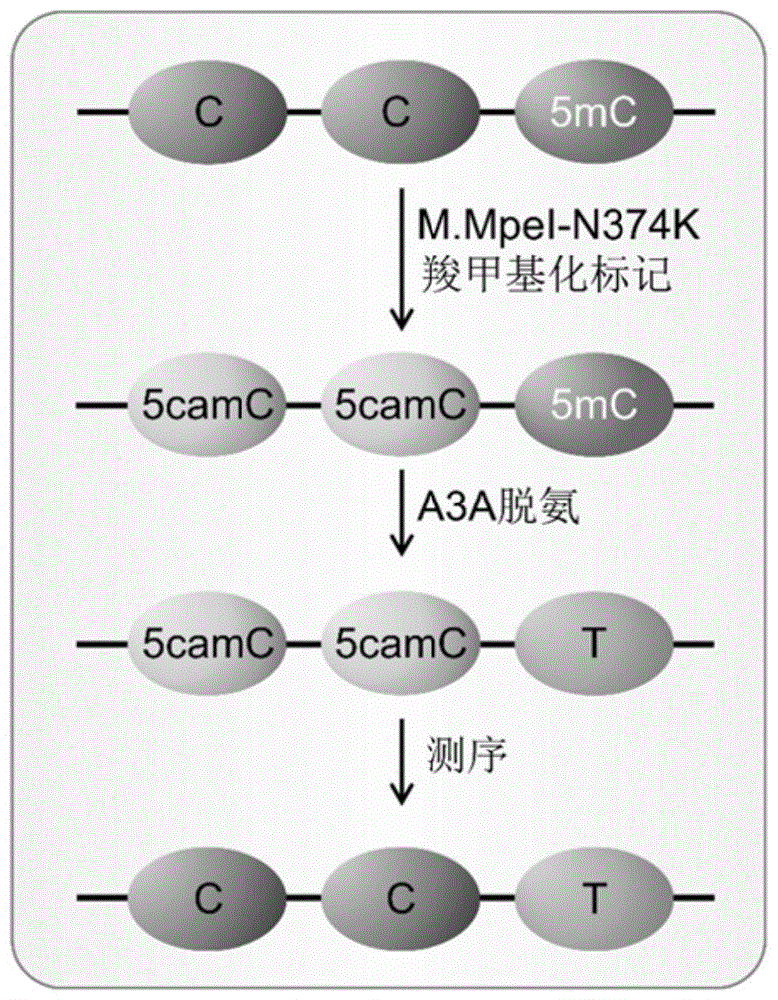
本发明提供的DNA甲基转移酶结合胞嘧啶脱氨酶介导的DNA中5-甲基胞嘧啶的单碱基定位分析方法,原理见图1,DNA甲基转移酶的突变体M.MpeI-N374K,以羧基-S-腺苷-L-甲硫氨酸为辅助因子,选择性地将羧甲基转移到CG二核苷酸中胞嘧啶的C5位,形成5-羧甲基胞嘧啶。在胞嘧啶脱氨酶A3A蛋白处理后,5-羧甲基胞嘧啶能够抵抗脱氨,因此在随后的聚合酶链式反应扩增中扩增为胞嘧啶,而目标分析物5mC无法被DNA甲基转移酶标记,且能够被A3A完全脱氨,在随后的聚合酶链式反应中扩增为胸腺嘧啶。因此,只有5-甲基胞嘧啶才能在最终的测序中被读为胸腺嘧啶。具备如下优点:
(1)本发明的方法方法操作简便,无需复杂的样品前处理;
(2)无需对DNA样品进行亚硫酸氢盐处理,反应条件温和,不会造成DNA的大量降解,可直接获得DNA中5-甲基胞嘧啶的单碱基分辨率定位信息;
(3)本发明DNA用量小,仅需100ng的基因组DNA即可对5mC进行定位分析。
(4)本发明无需TET蛋白氧化5mC,避免了TET氧化不完全造成假阴性结果的问题。
(5)本发明涉及的DNA甲基转移酶对转移羧甲基效率高(>90%),有利于5mC的准确分析。
(6)本发明涉及的胞嘧啶脱氨酶5mC具有高效的脱氨效率(97%),而对5camC的脱氨基效率很低(<0.1%),有利于5mC的定位分析,定位分析准确率高。
(7)本发明涉及的方法能够广泛应用于DNA中5mC修饰定位与定量的研究,并有助于对5mC的生物学功能进行更加深入的研究。
附图说明
为了更清楚地说明本发明实施例中的技术方案,下面将对实施例描述中所需要使用的附图作一简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其它的附图。
图1为本发明示意图。本发明中,C被M.MpeI-N374K标记上羧甲基,生成5camC,后者能够抵抗A3A蛋白的脱氨作用,而5mC不被标记且能够被A3A转化为T。因此,在测序时,只有5mC能够被读成T。
图2为本发明中利用变性聚丙烯酰胺凝胶电泳检测人工合成的DNA链(49-CG-FAM)被DNA甲基转移酶M.MpeI野生型(WT)与突变型(N374K)标记的效率。
图3A为本发明中利用LC-MS/MS检测人工合成的DNA链经羧甲基化标记后进行脱氨基处理前后5camC含量对比图。
图3B为本发明中利用LC-MS/MS检测人工合成的含有5mC修饰的DNA链经羧甲基化标记后进行脱氨基处理前后5mC含量对比图。
图4A为本发明中人工合成的不含5mC修饰的DNA链(DNA-C)和5mC修饰的DNA链(DNA-5mC)经M.MpeI-N374K羧甲基化标记与A3A蛋白处理后,Sanger测序的读出结果,其中DNA-C中的C经羧甲基化标记与A3A蛋白处理后均未突变,仍读作C;DNA-5mC中的5mC经羧甲基化标记与A3A脱氨后均突变为T。
图4B为不同比例的人工合成的不含5mC修饰的DNA链(DNA-C)和5mC修饰的DNA链(DNA-5mC)混合后,采用本发明中的方法测得的TCG位点处T/(C+T)峰高比值与理论5mC含量的相关性。
图4C为不同比例的人工合成的不含5mC修饰的DNA链(DNA-C)和5mC修饰的DNA链(DNA-5mC)混合后,采用本发明中的方法测得的CCG位点处T/(C+T)峰高比值与理论5mC含量的相关性。
图4D为不同比例的人工合成的不含5mC修饰的DNA链(DNA-C)和5mC修饰的DNA链(DNA-5mC)混合后,采用本发明中的方法测得的GCG位点处T/(C+T)峰高比值与理论5mC含量的相关性。
图4E为不同比例的人工合成的不含5mC修饰的DNA链(DNA-C)和5mC修饰的DNA链(DNA-5mC)混合后,采用本发明中的方法测得的ACG位点处T/(C+T)峰高比值与理论5mC含量的相关性。
图5A为通过本发明方法对人肺癌及癌旁组织DNA中RARB基因启动子的5mC进行单碱基分辨率检测。阴性对照是通过A3A蛋白对基因组DNA直接进行脱氨的结果。
图5B为通过本发明方法对肺癌旁和肺癌组织DNA的RARB基因启动子区域9个CG位点的5mC含量的定量结果。um,未甲基化。n.s.,无显著差异;*p<0.05(n=3);**p<0.01(n=3);***p<0.001(n=3)。
图5C为通过本发明方法和亚硫酸氢盐测序法对肺癌旁组织DNA的RARB基因启动子区域5mC含量的相关性分析。结果表明二者相关性良好,皮尔逊相关系数为0.937。
图5D为为通过本发明方法和亚硫酸氢盐测序法对肺癌组织DNA的RARB基因启动子区域5mC含量的相关性分析。结果表明二者相关性良好,皮尔逊相关系数为0.916。
图6为评价本发明方法和亚硫酸氢盐测序法中DNA的降解情况。将不同起始用量的DNA进行本发明方法和商品化亚硫酸氢盐方法(QIAGEN)处理后,进行PCR扩增,扩增产物在1.5%琼脂糖凝胶分离并检测。
具体实施方式
下文将结合具体实施方式和实施例,具体阐述本发明,本发明的优点和各种效果将由此更加清楚地呈现。本领域技术人员应理解,这些具体实施方式和实施例是用于说明本发明,而非限制本发明。
在整个说明书中,除非另有特别说明,本文使用的术语应理解为如本领域中通常所使用的含义。因此,除非另有定义,本文使用的所有技术和科学术语具有与本发明所属领域技术人员的一般理解相同的含义。若存在矛盾,本说明书优先。
除非另有特别说明,本发明中用到的各种原材料、试剂、仪器和设备等,均可通过市场购买得到或者可通过现有方法制备得到。
本发明实施例提供的技术方案为解决上述技术问题,总体思路如下:
根据本发明实施例一种典型的实施方式,提供一种DNA甲基转移酶结合胞嘧啶脱氨酶介导的DNA中5-甲基胞嘧啶的单碱基定位分析方法,所述方法包括:
S1、采用DNA甲基转移酶对待检DNA进行羧甲基化标记反应,后变性处理以使DNA甲基转移酶灭活而终止反应,获得羧甲基化标记的DNA;
所述步骤S1中,
所述羧甲基化标记反应的条件为:双链DNA在含Tris-HCl 5~20mM、pH 7.0~8.5、NaCl 20~100mM、二硫苏糖醇0.2~2mM、EDTA 0.2~2mM、羧基-S-腺苷-L-甲硫氨酸80~640mM、DNA甲基转移酶(M.MpeI-N374K蛋白)1~10μM的反应体系中,于30~45℃反应0.5~6小时。在具体的实施方式中,M.MpeI-N374K蛋白的浓度具体根据实际蛋白的活性来确定。所述羧甲基化标记反应的条件有利于保证DNA甲基转移酶的活性和羧甲基化标记反应的效率
所述变性处理的方法为:在0~60% DMSO存在下,将DNA在90~98℃高温下孵育2~30分钟。所述变性处理有利于后续的脱氨基反应。
作为优选地,所述DNA甲基转移酶为氨基酸序列如SEQ ID NO.1所示的M.MpeI-N374K蛋白。
在Mycoplasma penetrans中发现了一种CG特异性甲基转移酶M.MpeI。研究发现,M.MpeI的突变体(M.MpeI-N374K蛋白)可以将羧甲基从羧基-S-腺苷-L-甲硫氨酸(caSAM)转移到CG处的胞嘧啶,形成5-羧甲基胞嘧啶(5camC)。凭借5camC的独特性质,我们提出了一种无需亚硫酸氢盐处理、单碱基分辨率和定量检测5mC的方法,通过甲基转移酶标记结合A3A脱氨对DNA中的5mC进行测序。
S2、采用胞嘧啶脱氨酶对所述羧甲基化标记DNA进行脱氨基反应,后进行高温灭活处理以使胞嘧啶脱氨酶灭活而终止反应,获得脱氨基DNA样品;
所述步骤S2中,
所述脱氨基反应的条件为:DNA在含2-(N-吗啉)乙磺酸10~30mM、Triton X-1000.05~0.5%、pH 5.5~7.5、A3A蛋白1~10μM的反应体系中,于30~45℃反应0.5~4小时。选择上述成分组成的缓冲液的原因可以保证脱氨酶的活性和脱氨反应的效率。
所述高温灭活处理的条件包括:于92~98℃水浴中孵育5~20min。该反应条件有利于胞嘧啶脱氨酶灭活,从而有利于后续的PCR扩增过程。
作为优选的实施方式,所述的胞嘧啶脱氨酶为氨基酸序列如SEQ ID NO.2所示的A3A蛋白。
S3、将所述脱氨基DNA样品进行PCR反应,获得扩增产物;
所述PCR反应中需要使用与特异性引物对羧甲基保护及脱氨后的DNA进行扩增。扩增产物进行琼脂糖凝胶电泳,切胶纯化,进行Sanger测序。针对不同的待测DNA需要设计相对应的引物。
S4、将所述扩增产物测序,获得5-甲基胞嘧啶的位点信息。
若PCR扩增得到的是特定序列的DNA片段,扩增产物可以直接进行Sanger测序;若PCR扩增形成DNA文库,则将扩增产物进行高通量测序。
作为一种具体的实施方式,所述方法的具体操作步骤为:
(1)合成羧基-S-腺苷-L-甲硫氨酸。
(2)配制高浓度的M.MpeI-N374K反应缓冲液的储存液:Tris-HCl 100mM、pH 7.0~8.5、NaCl 500mM、二硫苏糖醇10mM、EDTA 10mM。
(3)配制高浓度的A3A反应缓冲液的储存液:200mM 2-(N-吗啉)乙磺酸(MES),1%的triton X-100,pH为5.5-7.5。
(4)表达并纯化M.MpeI-N374K蛋白。
(5)表达并纯化A3A蛋白。
(6)从生物样品中提取DNA,用RNaseA酶将DNA中的RNA去除。
(7)取100ng提取的DNA,超声进行片段化处理,片段化大小为300-500bp。
(8)将步骤(7)片段化后的DNA冷冻旋干,加入2μL水、0.5μL步骤(1)合成的羧基-S-腺苷-L-甲硫氨酸(终浓度80~640mM)、0.5μL步骤2配制的M.MpeI-N374K反应缓冲液和2μLM.MpeI-N374K蛋白,30~45℃下反应0.5~6小时。反应结束后,将样品加热至75~98℃变性2~30分钟使蛋白失活。
(9)在步骤(8)的DNA中加入2~12μL的DMSO,加水至16μL,在90~98℃水浴中孵育2~30分钟,随后立即转移至冰水浴中猝火。加入2μL步骤(1)配制的A3A反应缓冲液,加入2μL的A3A蛋白(终浓度1~10μM),30~45℃下反应0.5~4小时。反应结束后,将样品加热至75~98℃变性2~30分钟使蛋白失活。
(8)PCR反应,获得扩增产物,后测序并分析。
根据本发明实施例另一种典型的实施方式,提供了一种DNA甲基转移酶和胞嘧啶脱氨酶的组合在对DNA中5-甲基胞嘧啶进行单碱基分辨率定位的分析方法中的应用。
根据本发明实施例另一种典型的实施方式,提供了一种用于对DNA中5-甲基胞嘧啶进行单碱基分辨率定位的试剂盒,所述试剂盒包括:DNA甲基转移酶和胞嘧啶脱氨酶。
所述试剂盒还包括:
采用DNA甲基转移酶进行羧甲基化的反应体系:Tris-HCl 5~20mM、pH 7.0~8.5、NaCl 20~100mM、二硫苏糖醇0.2~2mM、EDTA 0.2~2mM、羧基-S-腺苷-L-甲硫氨酸80~640mM、M.MpeI-N374K蛋白1~10μM;
采用胞嘧啶脱氨酶进行脱氨基的反应体系:DNA在含2-(N-吗啉)乙磺酸10~30mM、Triton X-100 0.05~0.5%、pH 5.5~7.5、A3A蛋白1~10μM的反应体系中,于30~45℃反应0.5~4小时。
下面将结合实施例及实验数据对本申请的效果进行详细说明。若未特别指明,实施例中所用的技术手段包括核酸的提取、酶解以及聚合酶链式反应为本领域技术人员所熟知的常规手段。
实施例1.利用人工合成的DNA链区分胞嘧啶和5-甲基胞嘧啶
A.羧基-S-腺苷-L-甲硫氨酸的合成
为了利用DNA甲基转移酶M.MpeI-N374K进行羧甲基化标记,首先合成了羧甲基供体羧基-S-腺苷-L-甲硫氨酸(carboxy-S-adenosyl-L-methionine,caSAM)。合成方法为:将10mg S-腺苷-L-高半胱氨酸溶解于1.7mL 150mM碳酸氢铵水溶液中,混合后加入340mg碘乙酸,37℃下反应24小时。然后,加入12mL冰甲醇,-80℃中静置12小时,在4℃下以2000rpm离心30分钟,用冰甲醇洗涤两次,离心分离出沉淀物。最后再用液相色谱纯化,得到纯净的caSAM。
B.羧甲基化标记验证
为了验证本发明中DNA甲基转移酶M.MpeI-N374K的羧甲基化效率,选择了一条FAM标记的49bp DNA链与M.MpeI-N374K蛋白和caSAM反应,通过限制性酶酶切结合聚丙烯酰胺凝胶电泳验证反应效率。
(1)49bp DNA链序列
5′-AATTATTAAAAATATATAAAAC
3′-TTAATAATTTTTATATATTTTGG
(2)49bp DNA链进行羧甲基标记
反应总体积5μL,包含2μL 1μM的49bp DNA链、0.5μL步骤(1)合成的caSAM(终浓度320mM)、0.5μL M.MpeI-N374K反应缓冲液(Tris-HCl 100mM、pH 8.0、NaCl 500mM、二硫苏糖醇10mM、EDTA 10mM)和2μL M.MpeI-N374K蛋白(终浓度3.2μM),37℃下反应4小时。反应结束后,将样品加热至95℃变性5分钟使蛋白失活。
(3)限制性酶酶切结合聚丙烯酰胺凝胶电泳验证羧甲基化效率
HpaII(NEB)是一种对5mC敏感的限制性内切酶,能够特异性识别并切割CCGG序列,当CCGG中第二个C被5mC取代后,HpaII不能切割。同样地,当CCGG中第二个C被5-羧甲基胞嘧啶(5camC)取代后,HpaII也不能切割。因此,能够通过检测DNA链是否被保护不被HpaII切割来评估羧甲基化效率(图2)。
上述步骤(2)中反应后的DNA经缓慢退火恢复双链后,加入2μL HpaII反应缓冲液(NEB)和1μL HpaII(NEB),补加水至20μL,37℃反应1小时。反应结束后,将样品加热至95℃变性5分钟使蛋白失活。反应产物于20%变性聚丙烯酰胺凝胶电泳分离并检测。结果如图2所示,当以caSAM为羧甲基供体时,野生型M.MpeI没有羧甲基转移活性,而其突变体M.MpeI-N374K的所甲基化效率很高(>91%)。
C.液相色谱串联质谱分析5-羧甲基胞嘧啶和5-甲基胞嘧啶的脱氨效率
根据之前的研究,胞嘧啶脱氨酶A3A能够高效对C和5mC脱氨,而5mC的氧化产物,如5caC,能够强烈抑制脱氨。基于此,本发明验证了5-羧甲基胞嘧啶(5camC)的脱氨基活性。
(1)羧甲基化标记
反应所需DNA模板为未修饰和含5mC修饰的49bp DNA。
未修饰49bp DNA:
5′-AATTATTAAAAATATATAAAACCGGATTAAATATAATATAAATATAATT-3′/3′-TTAATAATTTTTATATATTTTGGCCTAATTTATATTATATTTATATTAA-5′。
5mC修饰49bp DNA:
5′-AATTATTAAAAATATATAAAAC
反应总体积5μL,包含1μL 1μM的未修饰49bp DNA、1μL 1μM的5mC修饰49bp DNA、0.5μL caSAM(终浓度320mM)、0.5μL M.MpeI-N374K反应缓冲液(Tris-HCl 100mM、pH 8.0、NaCl 500mM、二硫苏糖醇10mM、EDTA 10mM)和2μL M.MpeI-N374K蛋白(终浓度3.2μM),37℃反应4小时。反应结束后,将样品加热至95℃变性5分钟使蛋白失活。
(2)DNA链进行脱氨反应
DNA链中加入2μL的DMSO,加水至16μL,在95℃水浴中孵育10分钟,随后立即转移至冰水浴中猝火。加入2μL A3A反应缓冲液(200mM MES,1% Triton X-100,pH 6.5),加入2μLA3A蛋白(终浓度6μM),37℃反应2小时。反应结束后,将样品加热至95℃变性5分钟使蛋白失活。
(3)液相色谱串联质谱分析5camC和5mC的脱氨效率
将A3A处理后的DNA酶解为核苷,利用液相色谱串联质谱分析A3A处理前后5camC和5mC含量的变化情况。如图3A-3B所示,A3A处理后5camC含量没有显著降低,仍保留99.9%的5camC;而A3A处理后5mC含量显著降低,脱氨效率达到97%。该结果表明,A3A蛋白可以对5mC高效脱氨,但不能使5camC脱氨。
D.Sanger测序验证本发明方法用于区分C和5mC
A3A蛋白对5mC和5camC的差异性脱氨作用为我们提供了机会,为了对DNA中5mC进行单碱基分辨率分析,我们利用Sanger测序对本发明方法进行了考察。
(1)216bp DNA链的合成
为了进行Sanger测序,首先合成了一段216bp不含修饰及含5mC修饰的DNA链(分别命名为DNA-C和DNA-5mC),与A3A蛋白反应,通过质谱和Sanger测序验证脱氨率。
在50μL的反应体系中,加入0.5ng pUC19质粒DNA(包含目标扩增序列,购自擎科公司),5μL 10×反应缓冲液,2.5U Taq DNA聚合酶(Takara),2μL 10μM正向引物(序列为5′-AGTGACGCTGAGCTTGACGTCGCGC-3′),2μL 10μM反向引物(序列为5′-CCAACATTCCACTAACAATTACTCTCT-3′),1μL dATP、dGTP、TTP和dCTP(浓度均为2.5mM)。当合成5mC修饰的DNA链时,以5mdCTP取代dCTP即可。
聚合酶链式反应(PCR)程序为:(1)95℃变性2min;(2)95℃变性15s;(3)52℃退火20s;(4)72℃延伸30s;(2)-(4)步重复35次;72℃延伸5min。对PCR产物利用试剂盒进行回收纯化(Omega Bio-Tek Inc.,Norcross,GA,USA)。利用微量分光光度计对合成的DNA浓度进行定量。
合成的216bp DNA-C序列如SEQ ID NO.3所示;合成的216bp DNA-5mC序列如SEQID NO.4所示。
AGTGACGCTGAGCTTGACGTCGCGCGATGAGAGGTGATTATGAGTATGTATAGTGTTAGGAAGAGTGTAGTAATAGGATGAAGATGATTATATGATCGATGGTCCGTATGCGTAGAATACGTTGTTGTAGTGATTATAATGGAGTGAGAATGTAGATGAGTGGAGTAGGTAGTAAGATGTAGTGGTGACAGAGAGTAATTGTTAGTGGAATGTTGG(SEQID NO.3);
AGTGACGCTGAGCTTGACGTCGCGCGATGAGAGGTGATTATGAGTATGTATAGTGTTAGGAAGAGTGTAGTAATAGGATGAAGATGATTATATGAT
(2)人工合成的DNA链进行羧甲基化标记及脱氨反应
取10ng DNA-C或DNA-5mC进行羧甲基化反应,反应条件同步骤B(2)。随后进行A3A脱氨,脱氨条件同步骤C(2)。
(3)Sanger测序分析
在对2种人工合成的DNA链进行羧甲基化标记和A3A处理后,利用特异性引物(正向引物:5’-AGTGACGTTGAGTTTGACGTC-3’;反向引物:5’-CCAACATTCCACTAACAATTACTCTCTA-3’)进行PCR扩增。扩增程序为:95℃变性30s;35次循环包含95℃变性30s,52℃退火30s,68℃延伸1min;68℃延伸5min。扩增产物直接进行Sanger测序。
图4A的结果表明,在M.MpeI-N374K和A3A处理后,DNA-5mC中的所有5mC都被读作T。相反,DNA-C中CG位点的所有C在M.MpeI-N374K和A3A处理后被读作C。由于只有CG位点的C被M.MpeI-N374K羧甲基化,非CG位点的C(CCGG位点的第一个胞嘧啶)也被脱氨并读作T。这些结果表明,在本发明方法中,CG位点的C和5mC分别被读作C和T,表明本发明方法可以很好地区分DNA中CG位点的C和5mC。
接下来我们用本发明方法定量评估了单个CG位点的5mC水平。我们制备了各种DNA-C和DNA-5mC不同比例的混合物,DNA-5mC/(DNA-C+DNA-5mC)的摩尔百分比为0%、33%、66%和100%。在本发明中,序列中产生的C和T代表原始的C和5mC。因此,测量的T/(C+T)的比率可以表现出个别CG位点的5mC水平。如图4B-4E所示,Sanger测序结果显示,从原始TCG、CCG、GCG和ACG序列中测得的5mC水平与理论上的5mC百分比成正比(TCG:斜率=0.85,R
实施例2.人类基因组DNA特定位点中5mC的定量分析
我们接下来应用本发明方法对基因组DNA中特定位点的5mC水平进行量化。我们分析了视黄酸受体β(RARB)基因启动子区域的5mC水平,RARB是一种肿瘤抑制基因,已知可调节许多的生物过程。RARB基因启动子的高甲基化被认为是预测非小细胞肺癌(NSCLC)早期复发的重要因素。应用本发明方法进行基因组中5mC定位分析具体包括如下步骤:
A.收集一对NSCLC肺癌旁及肺癌组织,根据制造商的推荐程序,使用组织DNA试剂盒(Omega)提取基因组DNA。
B.使用超声波均质器(Scientz)将基因组DNA分割成平均大小为300-500bp的片段。
C.然后实施例1中的方法,用M.MpeI-N374K对100ng的片段化DNA进行羧甲基化标记和A3A脱氨。作为对照,直接对100ng的片段化DNA进行A3A脱氨。
D.脱氨后的DNA通过PCR进行扩增,扩增体系包含:
表1
RARB-F:5’-GGTTAGGAGGGTTTATTTTTTGTTAA-3’;
RARB-R:5’-AATCATTTACCATTTTCCAAACTTACT-3’。
扩增程序为:
表2
E.扩增产物直接进行Sanger测序。
F.为了对比亚硫酸氢盐测序法,采用商品化试剂盒(QIAGEN)进行亚硫酸氢盐脱氨处理,然后利用引物RARB-F和RARB-R进行PCR扩增,扩增产物进行Sanger测序。
我们采用本发明方法分析了RARB基因启动子Chr3:25,428,373-25,428,436区域CG位点的5mC水平(图5A)。本发明采用M.MpeI-N374K和caSAM作为辅助因子对来自肿瘤或肿瘤邻近的肺组织的基因组DNA进行羧甲基化,然后进行A3A处理和Sanger测序。为了排除其他DNA胞嘧啶修饰(如5caC)的干扰,采用未经M.MpeI-N374K处理的基因组DNA作为对照。结果显示,在A3A处理后,所有CG位点的C/5mC都被读作T(图5A)。接下来,我们采用本发明方法来定量CG位点的5mC水平(图5B)。定量结果表明,在RARB基因启动子的9个CG位点中,有7个位点的5mC水平在肺部肿瘤DNA中显著升高(P<0.05,图5B)。这些结果通过传统的亚硫酸盐测序得到进一步证实(图5C-5D)。利用本发明方法得到的5mC水平与亚硫酸氢盐测序得到的水平相当(Pearson's r=0.94和0.92,图5C-5D)。
由于常规的亚硫酸氢盐处理可能导致基因组DNA的严重降解,因此本应用例比较了使用本发明方法或亚硫酸氢盐测序的基因组DNA的降解情况。将片段化后的基因组DNA(300-500bp)进行本发明方法或亚硫酸氢盐测序分析。脱氨后,RARB基因启动子的247bp区域被PCR扩增。如图6,在亚硫酸氢盐测序法中,至少需要10ng的起始基因组DNA可以检测到247bp的扩增产物。然而,利用本发明方法,只需0.1ng的起始基因组DNA就能清楚地检测到扩增产物。这一结果表明,在只有有限的DNA样本的情况下,本发明方法优于亚硫酸氢盐测序法。
综上,本发明方法能够在有限的DNA样本中对区域DNA中CG位点的上5mC进行定量和单碱基分辨率检测,结合高通量测序,本发明方法应该能够实现CG位点5mC的全基因组测序。
最后,还需要说明的是,术语“包括”、“包含”或者其任何其他变体意在涵盖非排他性的包含,从而使得包括一系列要素的过程、方法、物品或者设备不仅包括那些要素,而且还包括没有明确列出的其他要素,或者是还包括为这种过程、方法、物品或者设备所固有的要素。
尽管已描述了本发明的优选实施例,但本领域内的技术人员一旦得知了基本创造性概念,则可对这些实施例作出另外的变更和修改。所以,所附权利要求意欲解释为包括优选实施例以及落入本发明范围的所有变更和修改。
显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。
- 脱氨酶介导的DNA中N4-甲基胞嘧啶的单碱基分辨率定位分析方法
- 人工改造脱氨酶辅助的DNA中5-甲基胞嘧啶修饰单碱基分辨率定位分析方法