当归指纹图谱及其检测方法
文献发布时间:2024-01-17 01:24:51
技术领域
本发明涉及一种中药的检测方法,具体涉及当归的指纹图谱及其检测方法。
背景技术
当归(Angelica sinensis(Oliv.)Diels)是伞形科植物多年生当归的干燥根,又名西归、秦归、云归,始载于《神农本草经》,为低温长日照植物,主产地为甘肃岷县。其生品性温,味甘、辛,归肝、心、脾经,具补血活血、调经止痛、润肠通便的功效,主治月经不调、经闭痛经,血虚萎黄、眩晕心悸等症,为临床常用的一味中药,明代李时珍《本草纲目》中有“当归调血,为女人要药”的记载。文献报道,当归主要含有苯肽类、有机酸及其酯类、糖类等成分,具有造血、抗炎、镇痛、抗血小板聚集等多方面药理作用。
中药指纹图谱作为一种综合的、可量化的鉴定手段,可用于评价中药材、中药制剂以及半成品的质量,是对中药整体质量控制的手段,能够系统、整体、专属地来表征中药的内在品质。现有技术中,关于当归特征图谱中化学成分的研究并不是很全面,尤其是当归中的洋川芎内脂Ⅰ和洋川芎内脂H等成分,由于两者为同分异构体,一直无法实现较好的分离。因此,为得到更全面的化学成分信息和更好的分离度,本发明建立了当归指纹图谱的检测方法。
发明内容
发明目的:本发明的目的是解决现有技术的不足,提供一种当归指纹图谱的检测方法,该检测方法可以客观、全面、准确的评价当归的的质量,对控制当归的质量和保证临床疗效具有重要意义。
为了实现上述目的,本发明采取的技术方案为:
本发明的第一个目的是提供一种当归指纹图谱的检测方法,所述方法包括以下步骤:
步骤1、对照品溶液的制备:
精密称取阿魏酸、藁本内酯、洋川芎内脂Ⅰ、洋川芎内脂H、洋川芎内脂A、绿原酸、阿魏酸松柏酯、丁烯苯酞对照品,用甲醇溶解,制成混标溶液;
步骤2、当归供试品溶液的制备:
取不同批次的当归药材,粉碎后加入甲醇超声处理,过微孔滤膜,得供试品溶液;
步骤3、分别精密吸取步骤1制备的对照品溶液和步骤2制备的当归供试品溶液,注入高效液相色谱仪,记录色谱图;
液相色谱条件为:
色谱柱:十八烷基硅烷键合硅胶色谱柱;
流动相:A相为乙腈,B相为体积百分比0.2%乙酸,梯度洗脱;
流速:1mL/min;
检测波长:280nm;
柱温:35℃;
进样量:10μL;
步骤4、将步骤3中获得的当归供试品溶液的色谱图导入中药色谱指纹图谱相似度评价系统;选择不同批当归的色谱图中均存在的色谱峰作为共有峰;用平均值计算法生成当归的对照指纹图谱,计算各共有峰的相对保留时间和相对峰面积;并根据对照品溶液色谱图的保留时间标注当归的对照指纹图谱中峰的化学成分,得到当归指纹图谱。
进一步的,步骤1中阿魏酸、藁本内酯、洋川芎内脂Ⅰ、洋川芎内脂H、洋川芎内脂A、绿原酸、阿魏酸松柏酯、丁烯苯酞对照品在混标溶液中的质量浓度为0.02mg/ml。
进一步的,步骤2中当归质量M与甲醇体积V的比为M:V=1g:(45~55)mL;所述超声的条件为:超声功率250W,频率40kHz,超声时间不低于30min;所述微孔滤膜孔径为0.22μm;优选的,当归质量M与甲醇体积V的比为M:V=1g:50mL。
进一步的,步骤3所述色谱柱为SHIMADZU Shim-pack GIST-C
进一步的,步骤3所述梯度洗脱程序如下表:
本发明的第二个目的是提供采用前述的建立方法得到的当归指纹图谱。
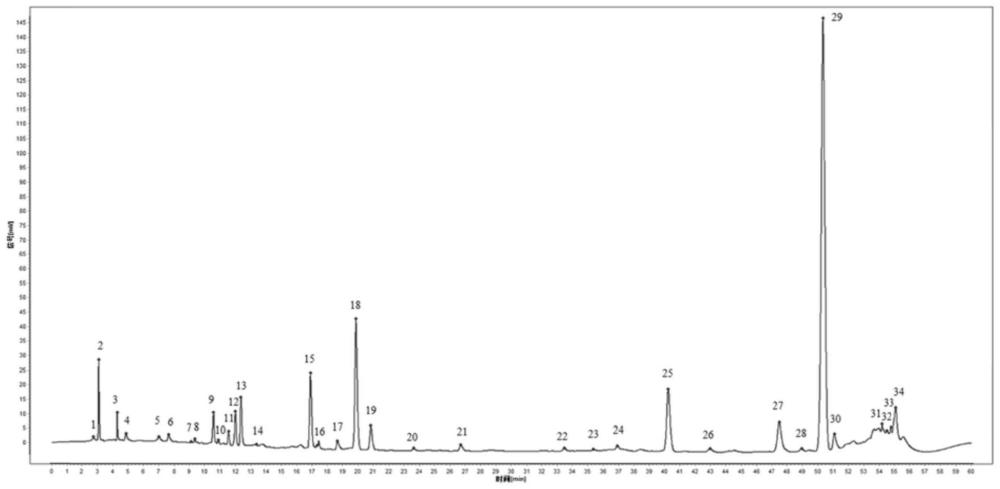
进一步的,所述指纹谱图中检测到34个共有峰。
进一步的,所述指纹谱图中识别8个成分,其中12号峰为绿原酸;15号峰为阿魏酸;18号峰为洋川芎内脂Ⅰ;19号峰为洋川芎内脂H;25号峰为阿魏酸松柏酯;26号峰为洋川芎内脂A;29号峰为藁本内酯;30号峰为丁烯苯酞。
进一步的,12号峰保留时间为11.659min;15号峰保留时间为16.523min;18号峰保留时间为19.477min;19号峰保留时间为20.437min;25号峰保留时间为39.808min;26号峰保留时间为42.581min;29号峰保留时间为49.995min;30号峰保留时间为50.827min。
本发明的第三个目的是提供前述的当归指纹图谱在当归质量控制中的应用。
指纹图谱检测条件的优化:
1、样品溶液的制备优化
本发明通过对不同提取方法(超声、加热回流)及不同提取溶剂(水、甲醇、无水乙醇、70%乙醇水溶液)进行实验比较,在不同提取方式的筛选中发现,加热回流提取和超声提取的样品的色谱图对比后,两种提取方式的色谱图差异较小,且所对应的成分峰都可清晰呈现,考虑到实验操作的便捷性和经济性,故采用超声提取的方法;对提取溶剂的考察中发现与其他提取溶剂相比,甲醇提取物色谱图信息量最多,成分含量最高,所以选用甲醇进行提取。
2、在色谱条件进行优化方面
本发明采用二极管阵列检测器对检测波长进行考察,发现检测波长为280nm时,色谱峰响应值比较高,被检测出来的成分最多,故选280nm为检测波长;
本发明比较了乙腈-水、乙腈-0.2%乙酸2个不同洗脱系统在不同梯度下的洗脱效果。结果发现以乙腈-0.2%乙酸为流动相时,当归中各成分能达到很好的分离效果,而流动相为乙腈-水时,各色谱峰峰型较差,分离度较低,故最终选定以乙腈-0.2%乙酸为流动相。
本发明又对洗脱程序进行了考察,发现等度洗脱不能实现良好的分离。又对梯度洗脱进行了大量的筛选,筛选得到最佳的梯度洗脱程序为0~8min,乙腈体积百分比5~25%;8~12min,乙腈体积百分比25~30%;12~25min,乙腈体积百分比30~40%;25~45min,乙腈体积百分比40~55%;45~50min,乙腈体积百分比55~90%;50~52min,乙腈体积百分比90~5%;52~60min,乙腈体积百分比5~5%。
有益效果
1、本发明根据当归中所含的活性成分的结构性质特点,通过大量实验筛选出最佳的供试品制备方法和色谱分析条件,建立的当归指纹图谱检测方法,可以全面、客观、准确的检测和评价当归的质量,为保证临床疗效具有重要意义。
2、本发明提供的当归纹图谱的检测方法,经方法学验证表明,具有稳定性好、精密度高、重现性好等优点。
附图说明
图1为对照品溶液的色谱图。
图2为当归样品的供试品对照指纹图谱。
图3为当归样品的20批次供试品指纹图谱。
具体实施方式
下面将结合实施例对本发明的实施方案进行详细描述,实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
实施例用到的仪器与试药如下:
仪器:日本岛津公司高效液相色谱仪,包括四元泵系统LC-20AT,全自动进样系统SIL-20A,自动温控柱温箱CTO-20AC;ML104/02电子分析天平(Mettler Toledo);XPR2电子分析天平(Mettler Toledo);KH-500E型超声波清洗器(昆山禾创超声仪器有限公司)。
试药:当归样品来源见表1;对照品阿魏酸(批号:0773-9910)、绿原酸(批号:110753-202018)均购于中国食品药品检定研究院;藁本内酯(批号:DH0007)、洋川芎内脂Ⅰ(批号:DY0009)、洋川芎内脂H(批号:DY0182)、洋川芎内脂A(批号:DY0008)、绿原酸(批号:110753-202018)、阿魏酸松柏酯(批号:DA0016)、丁烯苯酞(批号:101413-201601)购于成都德思特生物技术有限公司。甲醇(分析纯);乙腈(色谱纯);超纯水。
表1各批次药材的产地信息
实施例1一种当归指纹图谱的检测方法,包括以下步骤:
步骤1、对照品溶液的制备:
精密称定阿魏酸、藁本内酯、洋川芎内脂Ⅰ、洋川芎内脂H、洋川芎内脂A、绿原酸、阿魏酸松柏酯、丁烯苯酞对照品,加甲醇制成质量浓度分别为0.0279mg/ml、0.0172mg/ml、0.0259mg/ml、0.0192mg/ml、0.0175mg/ml、0.0262mg/ml、0.0194mg/ml、0.0290mg/ml的混合对照品溶液。
步骤2、当归供试品溶液的制备:
取20个批次当归药材粉末各1g,精密称定,置100ml具塞锥形瓶,精密加入50ml甲醇,密塞,称重,超声(功率250W,频率40kHz)处理30min,放冷至室温,再用甲醇补足损失的量,摇匀,经0.22μm的微孔滤膜过滤,即得。
步骤3、分别精密吸取上述对照品溶液和各批次供试品溶液,注入高效液相色谱仪,记录色谱图;液相色谱条件为:色谱柱:SHIMADZU Shim-pack GIST-C
混合对照品色谱图如图1所示,20批次供试品的指纹图谱见图3。
步骤4、将步骤3中获得的当归供试品溶液的指纹图谱导入中药色谱指纹图谱相似度评价系统2004A;选择不同批当归的色谱图中均存在的色谱峰作为共有峰;用平均值计算法生成当归的对照指纹图谱,计算各共有峰的相对保留时间和相对峰面积;并根据对照品溶液色谱图的保留时间标注对照指纹图谱中峰对应的化学成分。结果20批当归有34个共有峰,对照指纹图谱见图2。根据混合对照品确定12号峰为绿原酸,保留时间为11.659min;15号峰为阿魏酸,保留时间为16.523min;18号峰为洋川芎内脂Ⅰ,保留时间为19.477min;19号峰为洋川芎内脂H,保留时间为20.437min;25号峰为阿魏酸松柏酯,保留时间为39.808min;26号峰为洋川芎内脂A,保留时间为42.581min;29号峰为藁本内酯,保留时间为49.995min;30号峰为丁烯苯酞,保留时间为50.827min;。
用生成的对照指纹图谱R来建立共有色谱峰模式,分析计算获得不同批次的20批当归与共有模式之间相似度均在0.97以上,说明建立的指纹图谱可以很好的评价当归的质量,相似度结果见表2。
表2 20批次当归指纹图谱相似度结果
实施例2指纹图谱检测方法的方法学研究:
1、精密度考察
按实施例1方法制备的样品编号为S1的供试品溶液,按照实施例1的检测方法分析,平行进样6次,以藁本内酯色谱峰为参照峰,计算各共有峰的相对保留时间的RSD值均小于0.26%,相对峰面积的RSD值均小于2.03%,结果表明该设备的平行进样精密性良好。
2、重复性考察
平行精密称取编号为S1样品六份,按照实施例1的方法制成6份供试品溶液,参照实施例1的色谱条件,以藁本内酯色谱峰为参照峰,计算各共有峰的相对保留时间的RSD值均小于0.35%,相对峰面积的RSD值均小于2.81%,结果表明该方法的重复性良好。
3、稳定性考察
按实施例1方法制备的样品编号为S1的供试品溶液,按照实施例1的检测方法分析,采用0、2、4、8、12、24h不同时间进样分析,以藁本内酯色谱峰为参照峰,计算各共有峰的相对保留时间的RSD值均小于0.31%,相对峰面积的RSD值均小于2.67%,结果表明当归供试品溶液在24h之内的色谱峰几乎没有变化,稳定性较好。
以上实验结果表明,本发明提供的当归指纹图谱检测方法稳定性好,精密度高,重复性好,能全面客观评价当归药材的质量,为保证临床疗效具有重要的意义。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 当归饮片指纹图谱的建立方法及其指纹图谱
- 当归饮片指纹图谱的建立方法及其指纹图谱