一种酿酒酵母工程菌株及其应用
文献发布时间:2024-04-18 20:01:30
技术领域
本发明属于生物技术领域,具体涉及一种酿酒酵母工程菌株及其应用。
背景技术
二氢山奈酚广泛存在于豆类、西兰花、卷心菜、猕猴桃、葡萄、无头甘蓝、草莓、茶叶以及西红柿等植物性食物中,它具有抗氧化、抗菌、抗炎、降脂以及抗癌效应等多种功效,但价格昂贵。二氢山奈酚主要是从植物中分离纯化得到。传统植物化学法提取得到的产物多为二氢山奈酚混合物,下游纯化过程操作繁琐,对环境不友好。此外,由于这些植物中的二氢奈酚含量较低,且其生长的地理位置和气候条件存在差异,使用植物作为生产二氢山奈酚的来源会受制于其缓慢的生长速度和且组成成分并不均一,提取较为困难。。
黄烷酮-3-羟化酶(flavanone-3-hydroxylase,F3H)在黄酮类化合物的生物合成途径中发挥重要作用,其主要功能包括催化柚皮素C环第3位羟基化形成二氢山奈酚(dihydrokaempferol,DHK)。不同植物来源,如茶树(Camellia sinensis)、银杏(Ginkgobiloba)和大豆(Glycine max)、草莓(Fragaria x ananassa)、海棠(Malus x domestica)等的F3H具有相似的催化特性。相较于传统植物提取法,目前已有使用重组微生物细胞以更少的时间和能量消耗来合成获取二氢山奈酚的研究。例如,将不同植物来源的F3H基因在酿酒酵母中表达后,均可将柚皮素(Naringenin,NAR)转化为二氢山奈酚。但转化率较低,有待进一步提升。因此通过基因工程和代谢工程的手段改造酵母菌株以提高二氢山奈酚的产量及产率,具有重要的意义和实际的应用价值。
发明内容
本发明的目的在于提供能够显著提升将柚皮素转化为二氢山奈酚效率的酵母工程菌及其构建方法,解决现有酿酒酵母不能以柚皮素为底物,合成二氢山奈酚,及合成转化率低下的不足。通过发掘有效的长链非编码RNA(LncRNA)靶点,来提升酿酒酵母对柚皮素的转化能力,从而提高二氢山奈酚的产量及产率。本发明对表达F3H酿酒酵母的关键LncRNA靶点进行敲除,提升酵母细胞的柚皮素转化效率,克服了现有技术的缺陷,实现了二氢山奈酚合成能力的大幅提升。
本发明的目的通过下述技术方案实现:
一种酿酒酵母工程菌株,具有如下特点:LncRNA SUT067、LncRNA SUT433和LncRNACUT782中的至少一种缺失或突变;且含有能表达黄烷酮-3-羟化酶的编码核酸。
所述的酿酒酵母工程菌株的出发菌株优选为具有CEN.PK背景的菌株,该菌株是常用的酵母菌株,在工业界和学术界的代谢工程和系统生物学研究广泛应用(Microb CellFact,2012,11,36),代表性及普适性较好;优选为酿酒酵母CEN.PK 113-5D。
所述的缺失优选通过同源重组的方法或CRISPR/Cas9基因编辑技术进行无痕敲除。
所述的突变是使得相应的LncRNA不再发挥功能,可通过同源重组的方法或CRISPR/Cas9基因编辑技术得到。
所述的黄烷酮-3-羟化酶是植物来源的黄烷酮-3-羟化酶;优选为草莓黄烷酮-3-羟化酶和海棠黄烷酮-3-羟化酶。
所述的能表达黄烷酮-3-羟化酶的编码核酸,优选通过如下步骤得到:依据黄烷酮-3-羟化酶的氨基酸序列,进行适合酿酒酵母表达的密码子优化,将优化得到的编码黄烷酮-3-羟化酶的核酸克隆至表达载体中,得到能表达黄烷酮-3-羟化酶的编码核酸。
所述的黄烷酮-3-羟化酶为草莓黄烷酮-3-羟化酶时,优化的核酸序列优选如SEQID No.1所示。
所述的黄烷酮-3-羟化酶为海棠黄烷酮-3-羟化酶时,优化的核酸序列优选如SEQID No.2所示。
所述的表达载体优选为p416GPD、p416TEF、p416ADH、p416CYC1、p426GPD、p426TEF、p426ADH或p426CYC1。
所述的酿酒酵母工程菌株对柚皮素的转化效率高,从而其可用于制备二氢山奈酚。
一种二氢山奈酚的制备方法,包括如下步骤:
1)将上述酿酒酵母工程菌株接种于发酵培养基培养;
2)接着加入底物柚皮素,继续发酵培养,收集发酵产物即可得到二氢山奈酚。
步骤1)中所述的发酵培养基优选为YPD培养基。
步骤1)中所述的培养的条件优选为于28~32℃、150~250rpm培养20~30h;更优选为于30℃、200rpm培养24h。
步骤2)中所述的柚皮素的加入量优选按150~300mg/L计;更优选按200mg/L计。
步骤2)中所述的发酵培养的条件优选为于28~32℃、150~250rpm培养80~120h;更优选为于30℃、200rpm培养96h。
本发明相对于现有技术具有如下的优点及效果:
本发明对表达黄烷酮-3-羟化酶酵母工程菌进行了针对LncRNA靶点的改造,构建得到高产二氢山奈酚的工程菌对底物柚皮素转化率显著提升,均超过50.2%。其中,Q3M菌株转化率可达75.9%,相比对照菌株Q0M二氢山奈酚的产量提高了50%以上。不同于提高重组F3H表达质粒拷贝数等传统方法,该酵母工程菌仅从出发菌底盘细胞自身出发,围绕LncRNA相关靶点的敲除而展开。创新性地改造长非编码RNA靶点,有效的解决了酿酒酵母表达黄烷酮-3-羟化酶对底物转化率不高的首要问题。酵母工程菌用于提高其他高经济价值植物外源酶有较强的潜力,具备广阔的应用前景和重要的实践意义。
附图说明
图1是携带URA3筛选标记的重组表达质粒p416-FaF3H及p416-MdF3H的组成示意图。
图2是使用限制性内切酶SacI和BamHI对重组表达质粒p416-FaF3H及p416-MdF3H进行酶切验证的琼脂糖凝胶电泳图;其中,泳道M为DNA分子量marker。
图3是PCR鉴定敲除SUT067、SUT433、CUT782菌株的琼脂糖凝胶电泳图,其中泳道M为DNA分子量marker。
图4是PCR鉴定在ΔSUT067、ΔSUT433、ΔCUT782菌株中成功转入p416-FaF3H及p416-MdF3H质粒的琼脂糖凝胶电泳图;其中,泳道M为DNA分子量marker。
图5是实施例4中Q0F、Q1F、Q2F、Q3F酵母工程菌株试管发酵合成二氢山奈酚(DHK)的产量及底物柚皮素(NAR)转化率的数据图;其中,Q0F作为对照。
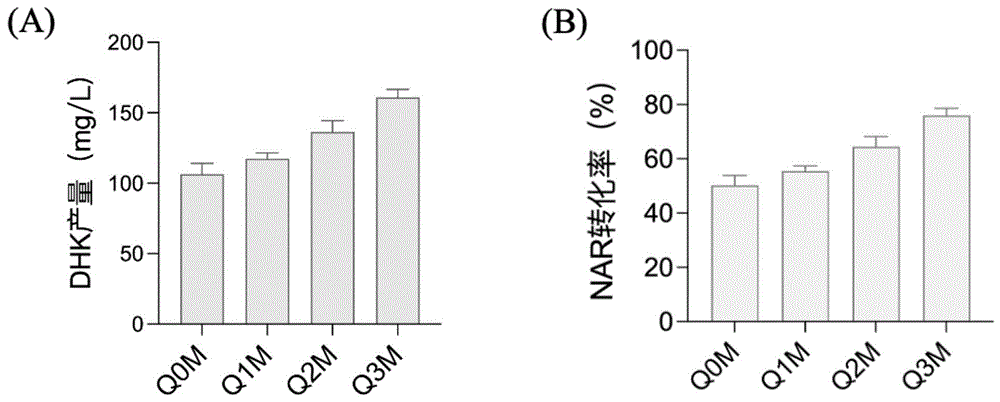
图6是实施例4中Q0M、Q1M、Q2M、Q3M酵母工程菌株试管发酵合成二氢山奈酚(DHK)的产量及底物柚皮素(NAR)转化率的数据图;其中,Q0M作为对照。
具体实施方式
下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到;
下列实施例中未注明具体条件的实验方法,则通常按照常规条件,如《分子克隆实验指南》(北京:科学出版社,2017)、《酵母遗传学方法实验指南》(北京:科学出版社,2016)中所述的条件进行。
下述实施例中应用到的CRISPR技术参见现有技术(Zhang et al.A gRNA-tRNAarray for CRISPR-Cas9based rapid multiplexed genome editing in Saccharomycescerevisiae.Nat Commun 2019,10(1):1053)。此外,下述实施例所使用的质粒psgtRNA、pCas质粒以及pScURA质粒片段的序列已在该文献“Zhang et al.AgRNA-tRNA array forCRISPR-Cas9 based rapid multiplexed genome editing in Saccharomycescerevisiae.Nat Commun 2019,10(1):1053”中的附件材料中公开,其中,pScURA质粒片段可通过人工合成即可得到。
为更好地理解本发明的内容,以酿酒酵母(Saccharomyces cerevisiae)CEN.PK背景菌株CEN.PK 113-5D(可由EUROSCARF获取)为出发菌株,为具体实施例作进一步说明。
下列实施例中所涉及的培养基如下:
LB液体培养基:10g/L蛋白胨,5g/L酵母提取物,10g/L NaCl,溶剂为去离子水;固体培养基添加2%琼脂粉。
LB/AMP培养基:10g/L蛋白胨,5g/L酵母提取物,10g/L NaCl,溶剂为去离子水;固体培养基添加2%琼脂粉;以上灭菌后冷却至40℃左右,加入100μg/mL氨苄青霉素(过滤除菌)。
YPD培养基:20g/L蛋白胨,10g/L酵母提取物,20g/L葡萄糖(单独灭菌后再加入),溶剂为去离子水;固体培养基添加2%琼脂粉。
SC-Ura营养缺陷培养基:0.77g/L CSM-Ura,1.7g/L YNB w/o AA&w/o(NH
下述实施例中所涉及的方法如下:
质粒构建:
(1)Gibson组装方法,具体操作依照NEB Gibson Assembly Cloning kit(货号E2611,NEB)说明书操作;
(2)Golden gate组装方法,具体操作依照
(3)将5μL组装体系转化至50μL大肠杆菌E.coli DH5α感受态细胞,涂布至LB/AMP固体培养基过夜培养;
(4)筛选获得阳性克隆,扩增培养后提取质粒,具体提取过程依照HiPure PlasmidMicro Kit(货号P1001-03,Magen)说明书操作。
酵母菌株转化:
如无特殊说明,均采用醋酸锂转化法,具体操作见相关标准规范。
重组酿酒酵母菌株OD
接种酿酒酵母单菌落于3mL发酵培养基,置于30℃、200rpm摇床培养。取发酵液按适当比例稀释后使用紫外分光光度计测量OD
二氢山奈酚检测方法:
柚皮素(NAR)及二氢山奈酚(DHK)标品购自Herbest(Baoji,China),含量测定时使用岛津高效液相色谱,Hypers11 ODS C18柱(250×4.6mm),检测波长290nm,流速1mL/min,柱温40℃洗脱程序如下表1:
表1
下述实施例中所涉及的引物序列如表2所示:
表2:引物序列(5’-3’)
实施例1草莓及海棠黄烷酮3-羟化酶编码基因的获取
根据UniProt数据库中公开的草莓(Fragaria ananassa)的黄烷酮3-羟化酶(Q66MF0)的氨基酸序列,海棠(Malus domestica)的黄烷酮3-羟化酶(Q06942)的氨基酸序列,并针对酿酒酵母进行密码子优化后由南京金斯瑞生物科技有限公司合成DNA编码序列(如SEQ ID No.1及SEQ ID No.2所示,皆编码364个氨基酸)并将其克隆于载体pUC57上,得到质粒pUC57-FaF3H及pUC57-MdF3H。
实施例2草莓黄烷酮3-羟化酶的表达载体的构建
草莓黄烷酮3-羟化酶的表达载体p416-FaF3H的结构如图1中的(A)所示,具体构建过程如下:
(1)使用引物p416-K-F/p416-K-R,以p416-GPD质粒为模板扩增质粒框架;PCR扩增包括常规的变性、退火以及延伸步骤(下同);其中退火温度为56℃,于72℃延伸300s,扩增30个循环。
(2)使用引物对Primer-A1F/Primer-A1R,以酿酒酵母CEN.PK 113-5D基因组为模板扩增出带有同源臂的PDC1启动子片段;PCR扩增的退火温度为56℃,于72℃延伸20s,扩增30个循环。
(3)使用引物对Primer-A2F/Primer-A2R,以pUC57-FaF3H为模板扩增出带有同源臂的FaF3H片段;PCR扩增的退火温度为56℃,72℃延伸20s,扩增30个循环。
(4)使用引物对Primer-A3F/Primer-A3R,以酿酒酵母CEN.PK 113-5D基因组为模板扩增出带有同源臂的PGK1终止子片段;PCR扩增的退火温度为56℃,72℃延伸20s,扩增30个循环。
(5)使用引物对FaF3H-F/FaF3H-R,以pUC57-FaF3H质粒为模板扩增出带有同源臂的FaF3H片段;PCR扩增的退火温度为56℃,72℃延伸60s,扩增30个循环。
(6)利用Gibson组装技术,将以上四种片段拼接起来,转化大肠杆菌DH5α。
经测序和酶切(通过SacI和BamHI酶切后的琼脂糖凝胶电泳图如图2所示)验证确认质粒构建成功,构建所得质粒命名为p416-FaF3H。
实施例3海棠黄烷酮3-羟化酶的表达载体的构建
海棠黄烷酮3-羟化酶的表达载体p416-MdF3H的结构如图1中的(B)所示,具体构建过程如下:
(1)使用引物p416-K-F/p416-K-R,以p416-GPD质粒为模板扩增质粒框架;PCR扩增的退火温度为56℃,于72℃延伸300s,扩增30个循环。
(2)使用引物对Primer-A1F/Primer-A1R,以酿酒酵母CEN.PK 113-5D基因组为模板扩增出带有同源臂的PDC1启动子片段;PCR扩增的退火温度为56℃,于72℃延伸20s,扩增30个循环。
(3)使用引物对Primer-A2F/Primer-A2R,以pUC57-MdF3H为模板扩增出带有同源臂的MdF3H片段;PCR扩增的退火温度为56℃,于72℃延伸20s,扩增30个循环。
(4)使用引物对Primer-A3F/Primer-A3R,以酿酒酵母CEN.PK 113-5D基因组为模板扩增出带有同源臂的PGK1终止子片段;PCR扩增的退火温度为56℃,于72℃延伸20s,扩增30个循环。
(5)使用引物对MdF3H-F/MdF3H-R,以pUC57-MdF3H质粒为模板扩增出带有同源臂的MdF3H片段;PCR扩增的退火温度为56℃,于72℃延伸60s,扩增30个循环。
(6)利用Gibson组装技术,将以上四种片段拼接起来,转化大肠杆菌DH5α。
经测序和酶切(通过SacI和BamHI酶切后的琼脂糖凝胶电泳图如图2所示)验证确认质粒构建成功,构建所得质粒命名为p416-MdF3H。
实施例4表达草莓黄烷酮3-羟化酶的酿酒酵母工程菌的构建
4.1敲除SUT067以高产二氢山奈酚
非编码RNA SUT067敲除过程如下:
(1)构建质粒pCas9-SUT067
非编码RNA SUT067的核苷酸序列如SEQ ID NO.3所示。
以质粒psgtRNA为模板,由引物对Primer-A4F/Primer-A4R扩增得到带有20bp靶向SUT067的gRNA序列,PCR扩增的退火温度为56℃,于72℃延伸20s,扩增30个循环。
以pScURA质粒片段为模板,由引物对Primer-A5F/Primer-A5R扩增得到1127bp带有营养标记URA3的质粒框架片段;PCR扩增的退火温度为56℃,于72℃延伸30s,扩增30个循环。
利用Golden gate组装技术,将以上两种片段与带有Cas9质粒片段的pCas质粒拼接起来,构建所得质粒命名为pCas9-SUT067。
(2)PCR扩增SUT067修复片段
合成与SUT067上下游完全匹配的59bp引物对Primer-A6F/Primer-A6R,经PCR扩增、凝胶回收获得与SUT067上下游有50bp同源臂的靶向修复片段,PCR扩增的退火温度为56℃,于72℃延伸20s,扩增30个循环。
(3)通过醋酸锂转化法,将质粒pCas9-SUT067连同SUT067修复片段一并转化至菌株CEN.PK 113-5D,涂布至SC-Ura固体培养基于30℃培养3~4天,使用引物对Primer-A7F/Primer-A7R进行单菌落PCR验证,筛选得到阳性转化子(结果图3所示),将质粒pCas9-SUT067去除,得到的酿酒酵母菌株命名为Q1。
转化质粒p416-FaF3H至菌株Q1以表达草莓黄烷酮3-羟化酶:
将酿酒酵母Q1制备成感受态细胞,并通过醋酸锂转化法将草莓黄烷酮3-羟化酶表达质粒p416-FaF3H转化至该感受态细胞,涂布至SC-Ura固体培养基上于30℃培养3-4天,使用引物对Primer-A8F/Primer-A8R进行单菌落PCR验证,筛选得到阳性菌株(结果图4所示),将其命名为酿酒酵母Q1F。
转化质粒p416-MdF3H至菌株Q1以表达海棠黄烷酮3-羟化酶:
将酿酒酵母Q1制备成感受态细胞,并通过醋酸锂转化法将海棠黄烷酮3-羟化酶表达质粒p416-MdF3H转化至该感受态细胞,涂布至SC-Ura固体培养基上于30℃培养3-4天,使用引物对Primer-A8F/Primer-A8R进行单菌落PCR验证,筛选得到阳性菌株(结果图4所示),将其命名为酿酒酵母Q1M。
4.2敲除SUT433以高产二氢山奈酚
非编码RNA SUT433敲除过程如下:
(1)构建质粒pCas9-SUT433
非编码RNA SUT433的核苷酸序列如SEQ ID NO.4所示。
以质粒psgtRNA为模板,由引物对Primer-A9F/Primer-A4R扩增得到带有20bp靶向SUT433的gRNA序列,PCR扩增的退火温度为56℃,72℃延伸20s,扩增30个循环。
以pScURA质粒片段为模板,由引物对Primer-A5F/Primer-A5R扩增得到1127bp带有营养标记URA3的质粒框架片段,PCR扩增的退火温度为56℃,72℃延伸30s,扩增30个循环。
利用Golden gate组装技术,将以上两种片段与带有Cas9质粒片段的pCas质粒拼接起来,构建所得质粒命名为pCas9-SUT433。
(2)PCR扩增SUT433修复片段
合成与SUT433上下游完全匹配的59bp引物对Primer-A10F/Primer-A10R,经PCR扩增、凝胶回收获得与SUT433上下游有50bp同源臂的靶向修复片段,PCR扩增的退火温度为56℃,72℃延伸20s,扩增30个循环。
(3)通过醋酸锂转化法,将质粒pCas9-SUT433连同SUT433修复片段一并转化至菌株CEN.PK 113-5D,涂布至SC-Ura固体培养基于30℃培养3-4天,使用引物对Primer-A11F/Primer-A11R进行单菌落PCR验证,筛选得到阳性转化子(结果图3所示),将质粒pCas9-SUT433去除,得到的酿酒酵母菌株命名为Q2。
转化质粒p416-FaF3H至菌株Q2以表达草莓黄烷酮3-羟化酶:
将酿酒酵母Q2制备成感受态细胞,并通过醋酸锂转化法将草莓黄烷酮3-羟化酶表达质粒p416-FaF3H转化至该感受态细胞,涂布至SC-Ura固体培养基上于30℃培养3-4天,使用引物对Primer-A8F/Primer-A8R进行单菌落PCR验证,筛选得到阳性菌株(结果图4所示),将其命名为酿酒酵母Q2F。
转化质粒p416-MdF3H至菌株Q2以表达海棠黄烷酮3-羟化酶:
将酿酒酵母Q2制备成感受态细胞,并通过醋酸锂转化法将海棠黄烷酮3-羟化酶表达质粒p416-MdF3H转化至该感受态细胞,涂布至SC-Ura固体培养基上于30℃培养3-4天,使用引物对Primer-A8F/Primer-A8R进行单菌落PCR验证,筛选得到阳性菌株(结果图4所示),将其命名为酿酒酵母Q2M。
4.3敲除CUT782以高产二氢山奈酚
非编码RNA CUT782敲除过程如下:
(1)构建质粒pCas9-CUT782
非编码RNA CUT782的核苷酸序列如SEQ ID NO.5所示。
以质粒psgtRNA为模板,由引物对Primer-A12F/Primer-A4R扩增得到带有20bp靶向CUT782的gRNA序列,PCR扩增的退火温度为56℃,72℃延伸20s,扩增30个循环。
以pScURA质粒片段,由引物对Primer-A5F/Primer-A5R扩增得到1127bp带有营养标记URA3的质粒框架片段PCR扩增的退火温度为56℃,72℃延伸30s,扩增30个循环。
利用Golden gate组装技术,将以上两种片段与带有Cas9质粒片段的pCas质粒拼接起来,构建所得质粒命名为pCas9-CUT782。
(2)PCR扩增CUT782修复片段
合成与CUT782上下游完全匹配的59bp引物对Primer-A13F/Primer-A13R,经PCR扩增、凝胶回收获得与CUT782上下游有50bp同源臂的靶向修复片段,PCR扩增的退火温度为56℃,72℃延伸20s,扩增30个循环。
(3)通过醋酸锂转化法,将质粒pCas9-CUT782连同CUT782修复片段一并转化至菌株CEN.PK 113-5D,涂布至SC-Ura固体培养基于30℃培养3-4天,使用引物对Primer-A14F/Primer-A14R进行单菌落PCR验证,筛选得到阳性转化子(结果图3所示),将质粒pCas9-CUT782去除,得到的酿酒酵母菌株命名为Q3。
转化质粒p416-FaF3H至菌株Q3以表达草莓黄烷酮3-羟化酶:
将酿酒酵母Q3制备成感受态细胞,并通过醋酸锂转化法将草莓黄烷酮3-羟化酶表达质粒p416-FaF3H转化至该感受态细胞,涂布至SC-Ura固体培养基上于30℃培养3-4天,使用引物对Primer-A8F/Primer-A8R进行单菌落PCR验证,筛选得到阳性菌株(结果图4所示),将其命名为酿酒酵母Q3F。
转化质粒p416-MdF3H至菌株Q3以表达海棠黄烷酮3-羟化酶:
将酿酒酵母Q3制备成感受态细胞,并通过醋酸锂转化法将海棠黄烷酮3-羟化酶表达质粒p416-MdF3H转化至该感受态细胞,涂布至SC-Ura固体培养基上于30℃培养3-4天,使用引物对Primer-A8F/Primer-A8R进行单菌落PCR验证,筛选得到阳性菌株(结果图4所示),将其命名为酿酒酵母Q3M。
实施例5工程菌株发酵生产二氢山奈酚
(1)分别将上述酿酒酵母菌株Q0F、Q1F、Q2F、Q3F,及Q0M、Q1FM、Q2M、Q3M从平板接种至2.5mL YPD发酵培养基,在30℃、200rpm条件下培养24h后,加入终浓度为200mg/L的柚皮素(NAR),其中Q0F及Q0M是对照菌。Q0F的制备过程如下:将酿酒酵母CEN.PK 113-5D制备成感受态细胞,并通过醋酸锂转化法将草莓黄烷酮3-羟化酶表达质粒p416-FaF3H转化至该感受态细胞,涂布至SC-Ura固体培养基上于30℃培养3-4天,使用引物对Primer-A8F/Primer-A8R进行单菌落PCR验证,筛选得到阳性菌株(结果图4所示),将其命名为酿酒酵母Q0F。Q0M的制备过程如下:将酿酒酵母CEN.PK 113-5D制备成感受态细胞,并通过醋酸锂转化法将海棠黄烷酮3-羟化酶表达质粒p416-MdF3H转化至该感受态细胞,涂布至SC-Ura固体培养基上于30℃培养3-4天,使用引物对Primer-A8F/Primer-A8R进行单菌落PCR验证,筛选得到阳性菌株(结果图4所示),将其命名为酿酒酵母Q0M。
(2)发酵96h后取发酵液稀释50倍后使用紫外分光光度计测量OD
(3)取1mL发酵液离心后取上清液600μL,加入600μL甲醇。
(4)震荡混匀混合液,离心后取500μL上清液,抽滤。
(5)测定二氢山奈酚含量以及计算转化率。
转化率=(二氢山奈酚产量*(柚皮素分子量/二氢山奈酚分子量)/添加柚皮素底物浓度)*100%。
表达草莓黄烷酮3-羟化酶的酵母的检测结果如图5中的(A)所示,可知Q1F、Q2F、Q3F菌株相比Q0F菌株的二氢山奈酚的产量(92.8mg/L)分别提高30.4%、30.2%、59.8%;图5中的(B)显示Q0F菌株对底物柚皮素转化率为43.8%,Q1F、Q2F菌株对底物转化率超过57%,Q3F则超过70%。
表达海棠黄烷酮3-羟化酶的酵母的检测结果如图6中的(A)所示,可知Q1M、Q2M、Q3M菌株相比Q0M菌株二氢山奈酚的产量(106.3mg/L)分别提高10.4%、28.2%、51.2%。图6中的(B)显示Q0M菌株对底物柚皮素转化率为50.2%,Q1M、Q2M菌株对底物转化率超过55%,其中,Q3M达到75.9%,具有广阔的产业化应用前景。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。