一种流感病毒未来突变的预测方法
文献发布时间:2024-04-18 20:01:55
技术领域
本发明涉及生物技术领域,具体涉及一种预测流感病毒突变的方法,进一步涉及一种流感病毒衣壳蛋白HA表面抗原区域的划定方法及在预测流感病毒突变中的作用。
背景技术
季节性流感病毒每年会感染全球5~15%的人口,并导致超过50万人死亡。作为一种典型的RNA病毒,季节性流感病毒不断地在表面衣壳蛋白,如血凝素 (HA)抗原上发生突变,以逃避之前由感染或疫苗接种所诱发的免疫力。由于HA 蛋白在宿主-病毒相互作用中的重要影响,目前它已被作为各种疫苗的主要成分。
每年,世界卫生组织(WHO)都会对现有疫苗的保护能力进行评估,如果保护效力明显下降,则会推荐新的疫苗成分。新的疫苗成分确定后,至少需要几个月的时间来进行疫苗的生产、包装和销售。这种明显的时间滞后可能导致几个月后接种的HA疫苗成分和流行的毒株不匹配,从而造成时不时的疫苗失效。
例如,在2004~2005年期间,世卫组织推荐的疫苗的有效性只有10%(95%置信区间[CI])。其原因是主要的社区流行毒株A/California/7/2004-like(H3N2)与 WHO推荐的疫苗毒株A/Fujian/411/2002-like(H3N2)之间的抗原性不一致,而后者在下一个流感季中已不再流行。同样的,2014~2015年的流感疫苗对占主导地位的A/H3N2流感病毒也没有提供有效保护,因为疫苗株 A/Texas/50/2012-like(H3N2)已经在人群中消失了。
事实上,疫苗从筛选到人群免疫之时间上的滞后性,不仅导致疫苗的失效,还导致疫苗有效性远低于预期。根据CDC的年度综合评估,2004~2020年的16 年间,流感疫苗的有效性在10%到60%之间波动,中位数仅为40.5%,这表明该领域还有很大的进步空间。换句话来说,由于病毒的不断快速变异,这种基于后验毒株的疫苗策略似乎无法对未来的突变毒株提供足够的保护。只有以流感未来变异做疫苗,疫苗效力才能得到有效提升。
持续的全球流感监测已累积了大量历史序列。许多进化研究聚焦于流感序列的溯源,而不是预测未来变异。预测季节性流感的未来抗原突变谱,有助于我们更好地制定流感的积极预防措施。
发明内容
本发明的目的在于提供一种有效预测流感未来突变的流感病毒衣壳蛋白 HA表面抗原区域的划定方法及流感病毒未来突变的预测方法,有助于提前开发预防性的治疗药物,以更好地应对流感威胁。
为实现上述目的,本发明采用的技术方案如下。
第一方面,本发明提供一种流感病毒衣壳蛋白HA表面抗原区域的划定方法。
一种流感病毒衣壳蛋白HA表面抗原区域的划定方法,包含以下步骤:
S101流感病毒数据集预处理:
收集公共数据库流感HA核酸序列集D,用多序列比对工具比对并截取HA 核苷酸序列中对应于HA1区域的第Ps×3-2位至Pe×3位,获得HA1核酸序列集 S,根据HA1核酸序列集S翻译蛋白序列集P;
其中,HA1为HA蛋白的一个结构域片段,Ps为HA1蛋白参考序列的起始位置,Pe为HA1蛋白参考序列的终止位置,不同亚型流感的HA参考序列HA1 的起始位置Ps和终止位置Pe不同(以influenza HA1 protein为关键词,在uniprot 数据库中检索不同亚型流感HA1蛋白的Ps、Pe的具体位点);
S102序列系统发育树构建:
基于时空转换原则构成代表序列集T,用以构建系统发育树并获得父-子节点序列集PT;
S103预测免疫原性区域划分:
根据公式MF
将包含所有高频突变位点的集合定义为高频突变位点集F;
优选地,将MF
优选地,将MF
从文献及数据库中收集实验验证的潜在免疫原性位点,获得实验位点集A;
定义F∪A为HA1潜在免疫原性位点集I,在HA1三维空间蛋白结构上,计算位点集I中各位点间距离矩阵D
根据聚类公式Cluster={c
优选地,所述的步骤S101中:所述的流感HA核酸序列集D可通过以下形式表示:D={d
所述的HA1核酸序列集S可通过以下形式表示:S={s
所述的翻译蛋白序列集P可通过以下形式表示:P={p
优选地,所述的步骤S102中,所述的时空转换原则为:时间为在每个流感季中,分别挑选N
优选地,构成的代表序列集T可通过以下形式表示:T={t
优选地,所述的步骤S103中:所述的D
优选地,所述的步骤S103中:所述的D
优选地,所述的步骤S103中:所述的D
优选地,所述的步骤S103中,对处在预测免疫原性区域C
第二方面,本发明提供一种流感病毒未来突变的预测方法。
一种流感病毒未来突变的预测方法,包含以下步骤:
S201免疫原性区域划分:根据本发明第一方面所述的方法将流感HA1蛋白中的高频突变位点和潜在免疫原型位点集合划分为n个预测免疫原性区域;
S202预测模板选择:
根据公式∑Ik(x=y)(I,k∈蛋白序列集P或核酸序列集S)计算HA1蛋白序列集P或HA1核酸序列集S中每条序列的丰度生成丰度降序集B;将已划分好的预测免疫原性区域根据映射关系F
S203模拟突变:
计算核苷酸突变频率,生成核苷酸突变概率矩阵Mn或氨基酸的突变频率生成氨基酸突变概率矩阵Mp;
所述的核苷酸突变概率矩阵Mn可通过以下方式表示:Mn={N
其中,M
当模板为核酸序列时,根据公式R=Mn×E进行突变模拟,最终获得潜在的突变谱R,所述的潜在的突变谱R可通过以下形式表示:R={r
当模板为蛋白序列时,根据公式R=Mp×E进行突变模拟,最终获得潜在的突变谱R,所述的潜在的突变谱R可通过以下形式表示:R={r
S204筛选及排序:
根据模拟的宿主免疫屏障公式:
根据公式∑Ik(x=y)(I,k∈潜在突变谱Q)计算潜在突变谱Q中每条子代序列的丰度,并按降序排序生成潜在突变谱U;或根据公式∑mut(mut∈潜在突变谱Q)统计每个突变在历史突变谱中被预测到的频次,并按降序排序生成潜在突变谱U,选择前N
在一个具体的实施例中,所述的步骤S202中:
所述的根据公式∑IK(x=y)中,当x=y时为1,否则为0;
所述的丰度降序集B可通过以下形式表示:B={b
所述的映射关系F
所述的模板集E为每个潜在免疫原性区域生成的模板集,可通过以下形式表示:E={e
在一个具体的实施例中,所述的步骤S204中:所述的潜在突变谱中每条子代序列突破免疫屏障的难易程度的衡量标准为,子代序列的DSIB>I(0.05~0.8) 时,剔除,经宿主免疫筛选后获得潜在突变谱Q;
所述的预测突变谱W的计算公式为W={w
所述的N
鉴于抗原区的高突变率,预测即将出现的突变对流感来说是非常具有技术挑战的研究。本发明提供的流感病毒衣壳蛋白HA表面新抗原区域的划定方法及流感病毒未来突变的预测方法,对抗原区域进行重新划定,并为每个流感季从流行毒株中获取多个模板,可以在更少的候选序列中捕获到更多的未来优势突变。
更重要的是,本发明的方法可以提前数个流感季预测到即将流行的新的优势突变。
本发明提供的方法可以根据目前不同亚型的流行毒株来预防流感威胁。
本发明提供的流感病毒未来突变的预测方法中,将筛选后的突变按模拟丰度进行排序,反映在社区中流行的潜在可能性。这种策略可以拓展至其他病毒。
综上所述,本发明提供了预测流感未来突变的可行性,并提出了一个实用的模型来解决这一难题。佐以抗原性评估,本发明可以有助于提前开发预防性的治疗药物或疫苗,以更好地应对流感威胁。
本发明提供的流感病毒衣壳蛋白HA表面新抗原区域的划定方法及流感病毒未来突变的预测方法的优点在于:
(1)能在更少的候选序列中捕获到更多的未来突变。
(2)能将潜在的优势突变排序靠前。
(3)能提前数年预测到未来在不同抗原区域出现的优势突变,从而为开发有针对性的流感预防措施节省时间。
附图说明
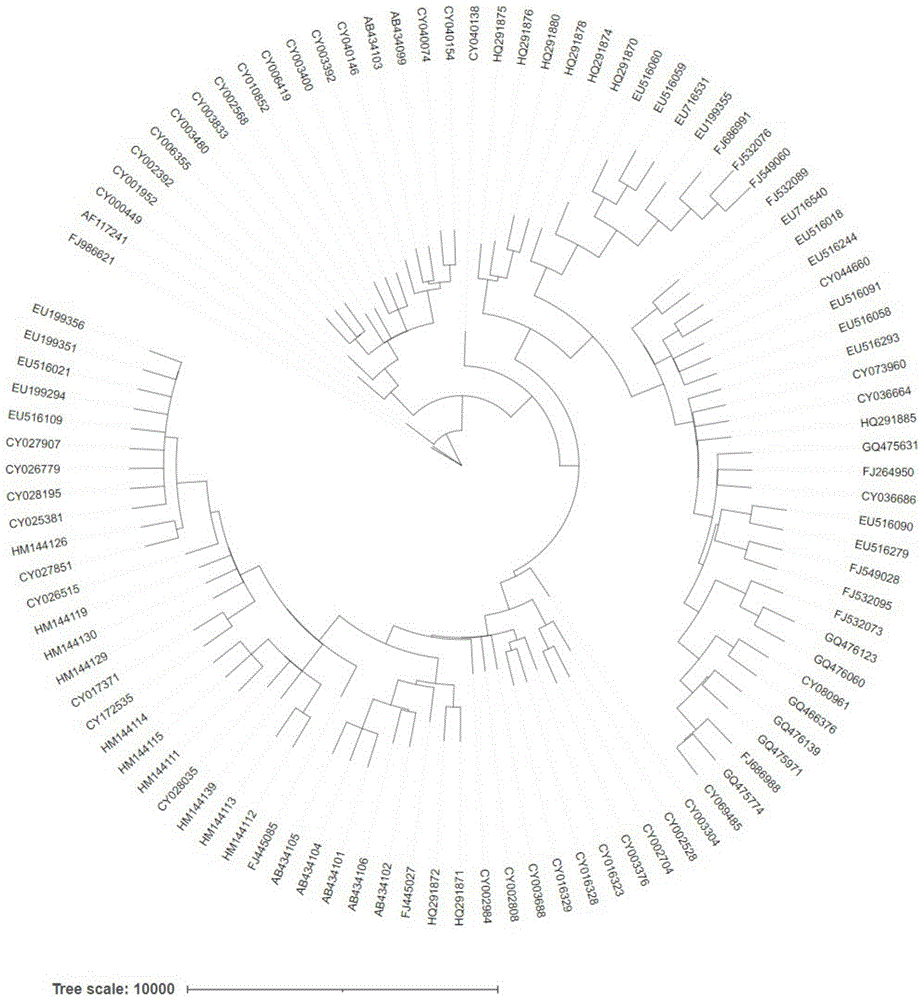
图1为本发明实施例1提供的H1N1_发育树。
图2为本发明实施例2提供的H3N2_发育树。
图3为本发明实施例3提供的H1N1预测性能(菌株覆盖率、实际观测到的突变以及被预测到的突变)结果。
图中,虚线代表每个抗原区域的菌株覆盖率,柱状图代表每个抗原区域实际观测到的突变数量,有灰度的部分代表被预测到的突变数量。不同字母代表不同抗原区域,A~G依次表示抗原区域A~G。
图4为本发明实施例3提供的H3N2预测性能(菌株覆盖率、实际观测到的突变以及被预测到的突变)结果。
图中,虚线代表每个抗原区域的菌株覆盖率,柱状图代表每个抗原区域实际观测到的突变数量,有灰度的部分代表被预测到的突变数量。不同字母代表不同抗原区域,A~F依次表示抗原区域A~F。
具体实施方式
为使本领域的技术人员更好地理解本发明的技术方案,以下实施例对本发明的作进一步详细描述,以下实施例仅用于说明发明,但不用来限制本发明的范围。
实施例1
一种H1N1型流感病毒衣壳蛋白HA表面抗原区域的划定方法,包含以下步骤:
收集公共数据库(收集自NCBI Influenza Virus Resources database)中的22919 条H1N1型流感的HA核酸序列,形成H1N1_HA核酸序列集D={AACAACCA…ATAAGGAAAAAA;AACATCTA…ATAAGGACCAAA;……;AACATCTA… ATAAGGACCAAA}(数据来源:NCBI Influenza Virus Resources database);
提交H1N1_HA核酸序列集D到多序列比对工具Cluster omega中进行多序列比对,形成H1N1_HA1核酸序列集S={AACATCCA…ATAAGGAATAAA; AACCTCTA…ATAAGGACCAAA;……;AACATCTT…ATAAGGACCAAT},(数据来源:NCBI Influenza VirusResources database);
根据H1N1_HA1核酸序列集S翻译形成H1N1_HA1蛋白序列集P={EDLPGNDNSTAT…NMRNVPEKQT;QDLPGNDNSTAT… GMRNVPEKQT;……QDLPGNDNSAAT…AMRNVPEKAT}(数据来源:NCBI Influenza Virus Resources database);
基于时空转换原则,时间为在每个流感季中,分别挑选N
表1H1N1代表序列集T
构建系统发育树,获得H1N1_HA1父-子节点序列集PT=AACATCCA…ATAAGGAATAAA:AACCTCTA…ATAAGGACCAAA;AACCTCTA…ATAAGGACCAAA:AACATCTT…ATAAGGACCAAT;……;AACATCTT… ATAAGGACCAAT:AACATCTT…ATAAGGACCAAT}(结果见图1);
根据公式MF
将包含所有高频突变位点的集合定义为高频突变位点集F,H1N1型流感 HA1蛋白序列的高频突变位点集F={35,36,43,54,69,71,82,84,85, 120,129,130,133,139,146,153,183,185,186,209,216,222,252,253,260,267,271,273,274,283,295,306,308,310};
根据Predicting the Mutating Distribution at Antigenic Sites of theInfluenza Virus(Scientific Reports,2016Feb 3|6:20239|DOI:10.1038/srep20239),收集经实验验证的免疫原性位点,形成H1N1型流感HA1蛋白序列的实验位点集A, A={73,74,75,76,77,78,127,128,140,141,142,143,144,145,156, 157,158,159,160,162,163,164,165,166,167,169,170,171,172, 173,187,188,189,190,191,192,193,194,195,196,197,198,206, 207,208,224,225,238,239,240},
定义H1N1_HA1潜在免疫原性位点集I=F∪A={35,36,43,54,69,71, 73,74,75,76,77,82,84,85,120,127,128,129,130,133,139,140, 141,142,143,144,145,146,153,156,157,158,159,160,162,163, 164,165,166,167,169,170,171,172,183,185,186,187,188,189, 190,191,193,194,195,196,198,206,207,208,209,216,222,224, 225,238,239,252,253,260,267,271,273,274,283,295,306,308,310};
基于HA1三维空间蛋白结构(H1N1:3m6s.pdb)计算H1N1_HA1潜在免疫原性位点集I中各位点氨基酸残基间的最小距离,形成矩阵D
根据聚类公式Cluster={c
对处在预测免疫原性区域边缘的位点,根据趋小原则i∈D
H1N1_HA1被划分为以下7个预测免疫原性区域:
AR A(35,36,85,267,283,295,306,308,310),
AR B(73,74,75,76,77,169,170,171,172,260),
AR C(43,54,69,71,82,84,271,273,274),
AR D(127,130,153,183,186,187,190,191,216,222,224,225),
AR E(156,157,158,159,160,185,188,189,193,194,195,196),
AR F(162,163,164,165,166,167,198,206,207,208,209,238, 239)
AR G(120,128,129,133,139,140,141,142,143,144,145,146,252,253)。
实施例2
一种H3N2型流感病毒衣壳蛋白HA表面抗原区域的划定方法,包含以下步骤:
收集公共数据库(收集自NCBI Influenza Virus Resources database)中的28183 条H3N2型流感的HA核酸序列,形成H3N2_HA核酸序列集D={AACAACCA…ATAAGGAAAAAA;AACATCTA…ATAAGGACCAAA;……;AACATCTA… ATAAGGACCAAA}(数据来源:NCBI Influenza Virus Resources database);
提交H3N2_HA核酸序列集D到多序列比对工具Cluster omega中进行多序列比对,形成H3N2_HA1核酸序列集S={AACATCCA…ATAAGGAATAAA; AACCTCTA…ATAAGGACCAAA;……;AACATCTT…ATAAGGACCAAT},(数据来源:NCBI Influenza VirusResources database);
根据H3N2_HA1核酸序列集S翻译形成H3N2_HA1蛋白序列集 P={EDLPGNDNSTAT…NMRNVPEKQT;QDLPGNDNSTAT… GMRNVPEKQT;……QDLPGNDNSAAT…AMRNVPEKAT}(数据来源:NCBI Influenza Virus Resources database);
基于时空转换原则,时间为在每个流感季中,分别挑选N
构建系统发育树,获得H3N2_HA1父-子节点序列集PT={AACATCCA…ATAAGGAATAAA:AACCTCTA…ATAAGGACCAAA;AACCTCTA… ATAAGGACCAAA:AACATCTT…ATAAGGACCAAT;……;AACATCTT… ATAAGGACCAAT:AACATCTT…ATAAGGACCAAT}(结果见图2);
根据公式MF
表2H3N2代表序列集T
将包含所有高频突变位点的集合定义为高频突变位点集F,H3N2型流感 HA1蛋白序列的高频突变位点集F={30,31,32,33,45,48,91,92,121, 128,135,138,140,142,159,165,167,171,173,198,210,212,225, 261,271,273,311,312};
根据Predicting the Mutating Distribution at Antigenic Sites of theInfluenza Virus(Scientific Reports,2016Feb 3|6:20239|DOI:10.1038/srep20239),收集经实验验证的免疫原性位点,形成H3N2型流感HA1蛋白序列的实验位点集A, A={50,53,54,62,75,82,83,122,124,131,133,137,143,144,145, 146,155,156,158,160,164,172,174,188,189,193,196,197,201, 207,213,217,230,244,260,262,276,278},
定义H3N2_HA1潜在免疫原性位点集I=F∪A={30,31,32,33,45,48, 50,53,54,62,75,82,83,91,92,121,122,124,128,131,133,135, 137,138,140,142,143,144,145,146,155,156,158,159,160,164, 165,167,171,172,173,174,188,189,193,196,197,198,201,207, 210,212,213,217,225,244,260,261,262,271,273,276,278,311, 312};
基于HA1三维空间蛋白结构(H3N2:4we8.pdb)计算H3N2_HA1潜在免疫原性位点集I中各位点氨基酸残基间的最小距离,形成矩阵D
根据聚类公式Cluster={c
对处在预测免疫原性区域边缘的位点,根据趋小原则i∈D
H3N2_HA1被划分为以下6个潜在免疫原性区域:
AR A(30,31,32,33,45,311,312),
AR B(82,83,121,122,124,171,172,173,174,260,261,262),
AR C(48,50,53,54,62,91,92,271,273,276,278),
AR D(131,155,156,158,159,160,188,189,193,196,197,198, 217),
AR E(128,165,164,167,201,207,210,212,213,244),
AR F(75,133,135,137,138,140,142,143,144,145,146,225)。
实施例3
预测H1N1流感病毒的未来突变,包含以下步骤:
免疫原性区域划分:根据实施例1所述的方法将H1N1型流感的HA1蛋白划分出7个潜在免疫原性区域;
预测模板选择:根据公式∑Ik(x=y)(I,k∈蛋白序列集P)计算HA1蛋白序列集P中每条序列的丰度,将已划分好的潜在免疫原性区域根据映射关系F
表3H1N1_模板集E
模拟突变:计算氨基酸的突变频率,生成氨基酸概率矩阵Mp={S->T:0.08, S->F:0.13,…,W->E:0.02},H1N1氨基酸概率矩阵Mp见表4。
筛选及排序:根据公式R=Mp×E进行突变模拟,并根据模拟的宿主免疫屏障公式:
H1N1突变预测性能结果见表6和图3。
表4H1N1_氨基酸概率矩阵Mp
/>
表5H1N1_预测突变谱W
/>
/>
表6H1N1预测性能结果
/>
/>
表中,
对H1N1,从2007年到2020年的13个流感季期间,每个流感季观测到5~63 种突变,本方法能在H1N1的7个抗原区域上平均预测到61%的突变类型(类型覆盖率),平均毒株覆盖率达到87%。
实施例4
预测H3N2流感病毒的未来突变,包含以下步骤:
免疫原性区域划分:根据实施例2所述的方法将H3N2型流感的HA1蛋白划分出6个潜在免疫原性区域;
预测模板选择:根据公式∑Ik(x=y)(I,k∈蛋白序列集P)计算H3N2_HA1蛋白序列集P中每条序列的丰度,将已划分好的潜在免疫原性区域根据映射关系 F
表7H3N2_模板集E
模拟突变:计算氨基酸的突变频率,生成氨基酸概率矩阵Mp={S->T:0.08, S->F:0.13,…,W->E:0.02},H3N2氨基酸概率矩阵Mp见表8;
筛选及排序:根据公式R=Mp×E进行突变模拟,并根据模拟的宿主免疫屏障公式:
表8H3N2-氨基酸概率矩阵Mp
表9H3N2_预测突变谱W
/>
H3N2突变预测性能结果见表10和图4。
表10H3N2预测性能结果
/>
/>
表中,
对H3N2,从2007年到2020年的13个流感季期间,每个流感季中观测到 3~67种突变,本方法能在H3N2的6个抗原区域上平均预测到63%的突变类型 (类型覆盖率),平均毒株覆盖率达到89%。
以上实施例的结果表明本方法可以精准预测每个流感季出现的大部分突变 (类型覆盖率),并且能预测到每个流感季中较为重要(流行度高)的突变(毒株覆盖率)。
实施例5
以本发明提供的流感病毒未来突变的预测方法与现有技术中的“突变-选择- 排序”的策略相对比结果如下:
预测性能的比较:
1.本发明提供的方法能在更少的候选序列中预测到更多的突变体
“突变-选择-排序”模型提供100条候选序列,2015~2019的平均strain coverage为87.8%,平均type coverage为37.8%;
本发明提供的方法提供90条候选序列,2015~2019的平均strain coverage 为97.1%,平均type coverage为66.3%。
与“突变-选择-排序”模型相比,本发明提供的方法的平均strain coverage 提升了10.6%,平均type coverage提升了75.4%。
2.本发明提供的方法对优势突变体(每个流感季中丰度最高的3个突变)的捕获率更高
“突变-选择-排序”模型中,2015~2019年期间,平均有69.9%的top3优势突变能被预测到;
本发明提供的方法中,2015~2019年期间,平均有93.8%的top3优势突变能被预测到。
与“突变-选择-排序”模型相比,对top3优势突变的平均捕获率提升了34.2%。
3.本发明提供的方法能预测未来新突变体(非上一流感季top3的突变体),而“突变-选择-排序”的策略不具备该性能
本发明提供的方法中,2015~2019年期间,新突变体的strain coverage平均为78.4%,新突变体的type coverage平均为62.3%。更重要的是,从现有数据看,本发明提供的方法中能提前2~8年预测到未来新优势突变,而“突变-选择-排序”模型不具备该预测性能。
其中,type coverage类型覆盖率:TC=T
strain coverage毒株覆盖率:SC=S
类型覆盖率和毒株覆盖率示例:
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种变换,这些简单变型均属于本发明的保护范围。
另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征和步骤,在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。
此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其同样应当视为本发明所公开的内容。