气相脱卤化氢制备3,3,3-三氟丙炔的方法
文献发布时间:2023-06-19 11:34:14
技术领域
本发明涉及一种气相脱卤化氢制备3,3,3-三氟丙炔的方法,尤其涉及一种以E-1-卤-3,3,3-三氟丙烯或/和Z-1-卤-3,3,3-三氟丙烯或/和2-卤-3,3,3-三氟丙烯(卤=F或Cl或Br或I)为原料,在催化剂存在下,经气相脱卤化氢反应,得到3,3,3-三氟丙炔的方法。
背景技术
目前,3,3,3-三氟丙炔的合成方法主要有以下两种:
(1)液相脱卤化氢
EP2143702报道了在乙醇溶剂中,130.5g的Z-1-氯-3,3,3-三氟丙烯与61.7g的氢氧化钾在38℃反应2.5小时,则Z-1-氯-3,3,3-三氟丙烯的转化率为99.2%,3,3,3-三氟丙炔的选择性为98.4%。
JP6032085报道了在氢氧化钾水溶液中,Z-1-氯-3,3,3-三氟丙烯在50℃反应2小时,反应压力不超过0.2MPa,则3,3,3-三氟丙炔的收率为85.9%,纯度为97.5%。
WO2016/132111报道了在高压釜中,Z-1-氯-3,3,3-三氟丙烯与40%质量百分比浓度的KOH水溶液在38℃反应5小时,则Z-1-氯-3,3,3-三氟丙烯的转化率为95.33%,3,3,3-三氟丙炔的选择性为99.8%。还报道了将溶解于甲醇(19.2 g)的Z-1233zd(20.4 g)装入装有冷凝器的100 mL高压釜中,该冷凝器与通过干燥冷却的样品瓶相连,将高压釜搅拌并加热至38℃,然后在2 h内通过注射泵添加40%KOH溶液(43.8 g),将混合物再加热3 h,并收集气态产物。 反应结果为:Z-1-氯-3,3,3-三氟丙烯的转化率为94.20%,,3,3,3-三氟丙炔的选择性为99.7%。
US2014/343330报道了在三颈烧瓶中,将氢氧化钾(粉末,31.6 g)和相转移催化剂Aliquat 336(化学式为C
文献“J. Med. Chem. 2011, 54, 9, 3393–3417”报道了在-78℃搅拌条件下,将二异丙胺溶解于四氢呋喃中,加入1.6M 的正丁基锂,维持温度低于-55℃,得到中间产物二异丙胺基锂,15分钟后,加入溶解于四氢呋喃的2-溴3,3,3-三氟-1-丙烯,维持温度低于-60℃,10分钟后,加入0.5 M氯化锌溶液,保持温度低于-65℃,继续将上述反应维持在78℃下搅拌10分钟,制备得到3,3,3-三氟丙炔,该反应属于定量反应。
文献“European Journal of Organic Chemistry,39(2020):6236-6244”报道了2,3,3,3-四氟丙烯在-78℃下于四氢呋喃中的正丁基锂反应1小时,然后与水反应,得到3,3,3-三氟丙炔。该反应属于定量反应。
文献“J. Chem. Soc.,(1951):588-591”报道了在带有液体空气冷凝装置的烧瓶中,1-碘-3,3,3-三氟丙烯在氢氧化钾存在下,油浴加热回流,油浴温度为120-150℃,回流时间6h,得到3,3,3-三氟丙炔,收率为70%。
文献“Journal of Fluorine Chemistry,12(1978):321-324”报道了在100 mL烧瓶中,以1,4-二氧己环为溶剂,1-碘-3,3,3-三氟丙烯在催化剂无水KF和助溶剂二环己烷并-18-冠醚-6存在下,加热回流3.5小时,则产物 3,3,3-三氟丙炔的收率为20%。
JP2018/90512报道了在连接压力表和排放阀的500 ml SUS-316反应器中,将16.80 g(0.3 mol)的氢氧化钾研磨粉和26.10 g(0.2 mol)的Z-1,3,3,3-四氟丙烯引入反应器并密封,将混合物在70℃加热9小时,同时用磁力搅拌器搅拌,反应的最终压力为0.5MPa,反应完成后,打开取出阀,将有机物液化到冷阱中(用甲醇+干冰冷却)并收集,反应结果为:Z-1,3,3,3-四氟丙烯的转化率为64.5%,3,3,3-三氟丙炔的选择性为98.4%。
上述液相法脱卤化氢的路线存在以下问题:(1)均属于间歇工艺,合成效率低下;(2)采用了大量的反应溶剂,会产生大量废液,从而严重污染环境;(3)使用了大量碱性的脱卤化氢试剂(如氢氧化钾、正丁基锂、二异丙基胺基锂等),会产生大量废固,从而严重污染环境;(4)采用正丁基锂或二异丙基胺基锂作为脱卤化氢的试剂,尽管3,3,3-三氟丙炔的收率很高,然而正丁基锂价格昂贵,使用条件苛刻,需要无水无氧操作,且易燃易爆,难以安全的实验操作。
(2)气相脱卤化氢
US2013/310614报道了在无催化剂存在下,2-氯-3,3,3-三氟丙烯在500℃、4.83MPa压力下发生脱氯化氢反应,2-氯-3,3,3-三氟丙烯流速为12g/h,则2-氯-3,3,3-三氟丙烯的转化率为29.1%,3,3,3-三氟丙炔的选择性为88.6%。
US2016/23972报道了在丸状活性炭(Shiarasagi生产,规格G2x4/16-1)存在下,2-氯-3,3,3-三氟丙烯在650℃发生脱氯化氢反应,接触时间30s,则2-氯-3,3,3-三氟丙烯的转化率为44.0%,3,3,3-三氟丙炔的选择性为56.8%。
WO2010/50373报道了在氟氧化铬催化剂(20.0 g,氟含量约为12.2 wt%)存在下,通入30mL/min的E-1,3,3,3-四氟丙烯进行反应,温度380℃,接触时间(催化剂质量与原料流速的比值)为40.0g-sec / mL,压力0.1MPa,连续运行2h后,反应结果为:E-1,3,3,3-四氟丙烯的转化率为40.7%,3,3,3-三氟丙炔的选择性为14.5%。
WO2010/95764报道了在KF/C催化剂(30g)存在下,通入20mL/min的E-1-氯-3,3,3-三氟丙烯进行反应,温度400℃,接触时间(催化剂质量与原料流速的比值)为60.0g-sec /mL,压力0.1MPa,连续运行2h后,反应结果为:E-1-氯-3,3,3-三氟丙烯的转化率为16.7%,3,3,3-三氟丙炔的选择性为83.2%。
上述气相法脱卤化氢的路线存在以下问题:催化剂的催化活性很低,表现出低转化率和低选择性的缺点,难以满足工业化生产的要求。
发明内容
本发明所要解决的技术问题是克服背景技术中存在的不足,提供一种单程产率较高、选择性较高的气相连续制备3,3,3-三氟丙炔的方法。
为了实现本发明的目的,本发明以E-1-卤-3,3,3-三氟丙烯或/和Z-1-卤-3,3,3-三氟丙烯或/和2-卤-3,3,3-三氟丙烯(卤=F或Cl或Br或I)为原料,通过气相脱卤化氢反应,高转化率、高选择性地合成3,3,3-三氟丙炔的方法,即:在催化剂存在下,在管式反应器中,E-1-卤-3,3,3-三氟丙烯或/和Z-1-卤-3,3,3-三氟丙烯或/和2-卤-3,3,3-三氟丙烯(卤=F或Cl或Br或I)发生气相脱卤化氢反应,得到3,3,3-三氟丙炔。反应方程式如下:
或/和
或/和
反应(1)至反应(3)中,X为F或Cl或Br或I。
本发明提供一种气相脱卤化氢制备3,3,3-三氟丙炔的方法,即:在催化剂存在下,在管式反应器中,1-卤-3,3,3-三氟丙烯或/和2-卤-3,3,3-三氟丙烯发生气相脱卤化氢反应,得到3,3,3-三氟丙炔,其中,卤为氟、氯、溴或碘,1-卤-3,3,3-三氟丙烯为Z-1-卤-3,3,3-三氟丙烯或/和E-1-卤-3,3,3-三氟丙烯;所述催化剂为碱土金属氟化物、或者由碱金属氟化物和碱土金属氟化物组成的催化剂,催化剂中碱金属氟化物和碱土金属氟化物质量百分比为0 ~ 30%和70% ~ 100%,所述碱金属氟化物为氟化锂、氟化钠、氟化钾、氟化铷、氟化铯中的任意一种或数种,碱土金属氟化物为氟化镁、氟化钡、氟化锶、氟化钡中的任意一种或数种,其中碱金属氟化物可以为零,也可以不为零。
催化剂组成中碱金属氟化物为零时,所述催化剂的其制备方法如下:(1)将碱土金属的可溶盐溶解于水,然后滴加沉淀剂,沉淀剂可为氨水或尿素中的任意一种,直至pH值为7 ~ 9,然后陈化10 ~ 24小时,过滤、洗涤,在50 ~ 120℃干燥10 ~ 24小时,得到固体,粉碎、压制成型,得到前驱体,其中,碱土金属的可溶盐为硝酸盐、氯化盐、醋酸盐或草酸盐;(2)所得前驱体,在氮气氛围下于300℃ ~ 500℃进行焙烧10 ~ 24小时;于200℃ ~ 400℃,在物质的量之比为1 :2的氟化氢与氮气组成的混合气体活化10 ~ 24小时,得到催化剂。
催化剂组成中碱金属氟化物不为零时,所述催化剂的其制备方法如下:(1)将碱土金属的可溶盐溶解于水,然后滴加沉淀剂,沉淀剂可为氨水或尿素中的任意一种,直至pH值为7 ~ 9,然后陈化10 ~ 24小时,过滤、洗涤,在50 ~ 120℃干燥10 ~ 24小时,得到固体,粉碎、压制成型,得到前驱体,其中,碱土金属的可溶盐为硝酸盐、氯化盐、醋酸盐或草酸盐;(2)所得前驱体,在氮气氛围下于300℃ ~ 500℃进行焙烧10 ~ 24小时;于200℃ ~400℃,在物质的量之比为1 :2的氟化氢与氮气组成的混合气体活化10 ~ 24小时,得到碱土金属氟化物;(3)按照碱金属氟化物和碱土金属氟化物的质量百分比组成,将碱金属氟化物溶解于水,向该浸渍液中加入碱土金属氟化物作为载体,室温下浸渍6 ~ 24小时,过滤,在50 ~ 120℃干燥10-24小时,在氮气氛围下于300℃ ~ 500℃进行焙烧10 ~ 24小时,得到催化剂。
所述气相脱卤化氢反应的反应条件为:反应压力0.1~0.5MPa,反应温度为300~600℃,1-卤-3,3,3-三氟丙烯或/和2-卤-3,3,3-三氟丙烯的接触时间为5~200s。
所述气相脱卤化氢反应的反应条件为:反应压力0.1~0.5MPa,反应温度为400~500℃,1-卤-3,3,3-三氟丙烯或/和2-卤-3,3,3-三氟丙烯的接触时间为30~150s。
所述1-卤-3,3,3-三氟丙烯为1-氯-3,3,3-三氟丙烯,2-卤-3,3,3-三氟丙烯为2-氯-3,3,3-三氟丙烯,所述气相脱卤化氢反应的反应条件为:反应压力0.1~0.5MPa,反应温度为400~500℃,1-氯-3,3,3-三氟丙烯或/和2-氯-3,3,3-三氟丙烯的接触时间为60~150s。
所述气相脱卤化氢反应的产物流为3,3,3-三氟丙炔、卤化氢、原料、以及原料的同分异构体产物组成的混合物,该混合物通过分离后,可以将卤化氢采出体系,原料以及原料的同分异构体产物循环至反应器继续反应,而分离得到的3,3,3-三氟丙炔经过后续的除酸、除水、精馏可以得到高纯度的产品3,3,3-三氟丙炔,其中,
当原料为E-1-卤-3,3,3-三氟丙烯,则产物流中原料的同分异构体产物为Z-1-卤-3,3,3-三氟丙烯和2-卤-3,3,3-三氟丙烯
当原料为Z-1-卤-3,3,3-三氟丙烯,则产物流中原料的同分异构体产物为E-1-卤-3,3,3-三氟丙烯和2-卤-3,3,3-三氟丙烯
当原料为2-卤-3,3,3-三氟丙烯,则产物流中原料的同分异构体产物为E-1-卤-3,3,3-三氟丙烯和Z-1-卤-3,3,3-三氟丙烯。
所述气相脱卤化氢工艺属于气相独立循环连续工艺方法。由于原料和反应产物的沸点差异很大,一般可以采用蒸馏塔蒸馏的方式将原料和产物进行有效分离,将未反应的原料以及与原料互为同分异构体的副产物继续循环至反应器继续参与反应,而产品3,3,3-三氟丙炔和副产品卤化氢采出体系。其中,2,3,3,3-四氟丙烯的沸点为-28℃(760mmHg);Z-1,3,3,3-四氟丙烯的沸点为9.8℃(760mmHg);E-1,3,3,3-四氟丙烯的沸点为-18.95℃(760mmHg);2-氯-3,3,3-三氟丙烯的沸点为12℃(760mmHg);Z-1-氯-3,3,3-三氟丙烯的沸点为40℃(760mmHg);E-1-氯-3,3,3-三氟丙烯的沸点为19.4℃(760mmHg);2-溴-3,3,3-三氟丙烯的沸点为34℃(760mmHg);E-1-溴-3,3,3-三氟丙烯的沸点为40℃(760mmHg);2-碘-3,3,3-三氟丙烯的沸点为56℃(760mmHg);E-1-碘-3,3,3-三氟丙烯的沸点为70.5℃(760mmHg);3,3,3-三氟丙炔的沸点为-48℃(760mmHg);氟化氢的沸点为19.5℃(760mmHg);氯化氢的沸点为-85.05℃(760mmHg);溴化氢的沸点为-66.38℃(760mmHg);碘化氢的沸点为-35.36℃(760mmHg)等等。
本发明用于反应的反应器类型不是关键,可以使用管式反应器,流化床反应器等。另外,绝热反应器或等温反应器亦可用。
本发明的优点:
(1)本发明的原料易得。其中原料Z-1-氯-3,3,3-三氟丙烯、E-1-氯-3,3,3-三氟丙烯、2-氯-3,3,3-三氟丙烯、Z-1,3,3,3-四氟丙烯、E-1,3,3,3-四氟丙烯、2,3,3,3-四氟丙烯可以从市场上直接购买获得;原料E-1-碘-3,3,3-三氟丙烯按“J. Chem. Soc.,(1951):588-591”文献方法进行制备;原料Z-1-碘-3,3,3-三氟丙烯按“Journal of the ChemicalSociety, Faraday Transactions 1: Physical Chemistry in Condensed Phases, 77(1981):89-100”文献方法进行制备;原料2-碘-3,3,3-三氟丙烯“Chemistry - A EuropeanJournal,4(1998) :1799–1809” 文献方法进行制备;原料E-1-溴-3,3,3-三氟丙烯或Z-1-溴-3,3,3-三氟丙烯按“J. Chem. Soc.,(1951) :2495-2504” 文献方法进行制备;原料2-溴-3,3,3-三氟丙烯“Chemistry - A European Journal,4(1998) :1799–1809” 文献方法进行制备。
(2)本发明合成3,3,3-三氟丙炔的单程产率较高、选择性较高;与WO2010/50373(E-1,3,3,3-四氟丙烯的转化率为40.7%,3,3,3-三氟丙炔的选择性为14.5%)的技术相比,本发明的单程产率更高、选择性更高(见实施例14:E-1,3,3,3-三氟丙烯的转化率为46.8%,3,3,3-三氟丙炔的选择性为30.1%);与WO2010/95764(E-1-氯-3,3,3-三氟丙烯的转化率为16.7%,3,3,3-三氟丙炔的选择性为83.2%)的技术相比,本发明的单程产率更高、选择性更高(见实施例12:E-1-氯-3,3,3-三氟丙烯的转化率为22.3%,3,3,3-三氟丙炔的选择性为96.1%)。
(3)本发明采用气相法制3,3,3-三氟丙炔,通过气相独立循环工艺,将反应不完全的物料进行独立循环,可以使初始原料几乎完全地转化目标产物,最终从工艺体系中采出目标产品和副产品卤化氢,从而不产生液废和废气,实现绿色生产。
附图说明
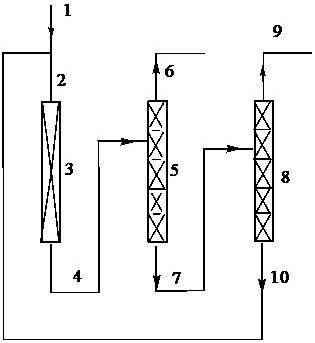
图1表示Z-1-氯-3,3,3-三氟丙烯为原料制备3,3,3-三氟丙炔的制备工艺流程图。
在图1中的标号意义如下。管线:1、2、4、6、7、9和10;反应器:3;第一蒸馏塔:5;第二蒸馏塔:8。
具体实施方式
下面结合附图说明对本发明做进一步详细说明。
参照图1对本发明进一步详细说明。但并不限制本发明。新鲜的Z-1-氯-3,3,3-三氟丙烯经管线1与经管线10的循环使用的Z-1-氯-3,3,3-三氟丙烯、E-1-氯-3,3,3-三氟丙烯和2-氯-3,3,3-三氟丙烯的混合物一起经过管线2进入装填有催化剂的反应器3进行气相脱卤化氢反应,反应产物流为3,3,3-三氟丙炔、Z-1-氯-3,3,3-三氟丙烯、E-1-氯-3,3,3-三氟丙烯、2-氯-3,3,3-三氟丙烯和氯化氢,反应产物流经管线4进入第一蒸馏塔5进行分离;第一蒸馏塔5的塔顶组分为氯化氢,塔釜组分为3,3,3-三氟丙炔、Z-1-氯-3,3,3-三氟丙烯、2-氯-3,3,3-三氟丙烯和E-1-氯-3,3,3-三氟丙烯,塔顶组分经管线6采出体系,可以继续精馏、除水得到高纯度HCl进行售卖,也可以配置成不同浓度的盐酸进行售卖;第一蒸馏塔5的塔釜组分经管线7进入第二蒸馏塔8继续分离;第二蒸馏塔8的塔顶组分是3,3,3-三氟丙炔,经管线9导出,再经过后续的除酸、除水、精馏可以得到高纯度的产品3,3,3-三氟丙炔;第二蒸馏塔8的塔釜组分是Z-1-氯-3,3,3-三氟丙烯、2-氯-3,3,3-三氟丙烯和E-1-氯-3,3,3-三氟丙烯,经管线10和管线2循环至反应器3继续反应。
分析仪器:岛津GC-2010,色谱柱型号为InterCap1 (i.d. 0.25 mm; length 60m; J&W Scientific Inc .)。
气相色谱分析方法:高纯氦气和氢气用作载气。检测器温度240℃,汽化室温度150℃,柱初温40℃,保持10分钟,20℃/min升温至240℃,保持10分钟。
实施例1
催化剂的制备:(1)将氯化钡溶解于水,然后滴加氨水,直至pH值为7-9,然后陈化16小时,过滤、洗涤,在100℃干燥16小时,得到固体,粉碎、压制成型,得到前驱体氢氧化钡;(2)所得前驱体,在氮气氛围下于400℃进行焙烧12小时;于300℃,在物质的量之比为1 :2的氟化氢与氮气组成的混合气体活化16小时,得到氟化钡;(3)按照氟化铯和氟化钡的质量百分比组成10%:90%,将碱金属氟化物溶解于水,向该浸渍液中加入氟化钡载体,室温下浸渍12小时,过滤,在100℃干燥16小时,在氮气氛围下于400℃进行焙烧12小时,得到催化剂。
在内径1/2英寸、长30cm的因康合金制的管式反应器中装填10毫升上述制备的催化剂。反应器升温至400℃,通入Z-1-氯-3,3,3-三氟丙烯进行反应,接触时间为100秒,反应压力为常压,反应20h后,反应产物经水洗、碱洗,分离得到有机物,经干燥除水后,用气相色谱分析有机物的组成,Z-1-氯-3,3,3-三氟丙烯的转化率为23.1%,3,3,3-三氟丙炔的选择性为95.6%,E-1-氯-3,3,3-三氟丙烯和2-氯-3,3,3-三氟丙烯的选择性之和为4.0%。
实施例2
与实施例1相同的操作,所不同的是“按照氟化铯和氟化钡的质量百分比组成10%:90%”改为“按照氟化铯和氟化钡的质量百分比组成20%:80%”,将反应温度改为350℃。反应结果如下:Z-1-氯-3,3,3-三氟丙烯的转化率为13.8%,3,3,3-三氟丙炔的选择性为96.8%,E-1-氯-3,3,3-三氟丙烯和2-氯-3,3,3-三氟丙烯的选择性之和为3.1%。
实施例3
与实施例1相同的操作,所不同的是“按照氟化铯和氟化钡的质量百分比组成10%:90%”改为“按照氟化铯和氟化钡的质量百分比组成30%:70%”,将反应温度改为450℃。反应结果如下:Z-1-氯-3,3,3-三氟丙烯的转化率为25.4%,3,3,3-三氟丙炔的选择性为93.1%,E-1-氯-3,3,3-三氟丙烯和2-氯-3,3,3-三氟丙烯的选择性之和为6.8%。
实施例4
催化剂的制备:(1)将氯化钡溶解于水,然后滴加氨水,直至pH值为7-9,然后陈化16小时,过滤、洗涤,在100℃干燥16小时,得到固体,粉碎、压制成型,得到前驱体氢氧化钡;(2)所得前驱体,在氮气氛围下于400℃进行焙烧12小时;于300℃,在物质的量之比为1 :2的氟化氢与氮气组成的混合气体活化16小时,得到氟化钡催化剂。
在内径1/2英寸、长30cm的因康合金制的管式反应器中装填10毫升上述制备的催化剂。反应器升温至500℃,通入Z-1-氯-3,3,3-三氟丙烯进行反应,接触时间为100秒,反应压力为常压,反应20h后,反应产物经水洗、碱洗,分离得到有机物,经干燥除水后,用气相色谱分析有机物的组成,Z-1-氯-3,3,3-三氟丙烯的转化率为26.8%,3,3,3-三氟丙炔的选择性为91.2%,E-1-氯-3,3,3-三氟丙烯和2-氯-3,3,3-三氟丙烯的选择性之和为8.6%。
实施例5
与实施例1相同的操作,所不同的是将接触时间改为5秒。反应结果如下:Z-1-氯-3,3,3-三氟丙烯的转化率为15.8%,3,3,3-三氟丙炔的选择性为96.2%,E-1-氯-3,3,3-三氟丙烯和2-氯-3,3,3-三氟丙烯的选择性之和为3.8%。
实施例6
与实施例1相同的操作,所不同的是将接触时间改为30秒。反应结果如下:Z-1-氯-3,3,3-三氟丙烯的转化率为18.3%,3,3,3-三氟丙炔的选择性为95.9%,E-1-氯-3,3,3-三氟丙烯和2-氯-3,3,3-三氟丙烯的选择性之和为4.1%。
实施例7
与实施例1相同的操作,所不同的是将接触时间改为60秒。反应结果如下:Z-1-氯-3,3,3-三氟丙烯的转化率为21.4%,3,3,3-三氟丙炔的选择性为95.7%,E-1-氯-3,3,3-三氟丙烯和2-氯-3,3,3-三氟丙烯的选择性之和为4.3%。
实施例8
与实施例1相同的操作,所不同的是将接触时间改为150秒。反应结果如下:Z-1-氯-3,3,3-三氟丙烯的转化率为23.8%,3,3,3-三氟丙炔的选择性为93.4%,E-1-氯-3,3,3-三氟丙烯和2-氯-3,3,3-三氟丙烯的选择性之和为6.5%。
实施例9
与实施例1相同的操作,所不同的是将接触时间改为200秒。反应结果如下:Z-1-氯-3,3,3-三氟丙烯的转化率为24.2%,3,3,3-三氟丙炔的选择性为92.1%,E-1-氯-3,3,3-三氟丙烯和2-氯-3,3,3-三氟丙烯的选择性之和为7.8%。
实施例10
与实施例1相同的操作,所不同的是反应压力改为0.3MPa。反应结果如下:Z-1-氯-3,3,3-三氟丙烯的转化率为21.4%,3,3,3-三氟丙炔的选择性为94.3%,E-1-氯-3,3,3-三氟丙烯和2-氯-3,3,3-三氟丙烯的选择性之和为5.7%。
实施例11
与实施例1相同的操作,所不同的是反应压力改为0.5MPa。反应结果如下:Z-1-氯-3,3,3-三氟丙烯的转化率为20.3%,3,3,3-三氟丙炔的选择性为92.1%,E-1-氯-3,3,3-三氟丙烯和2-氯-3,3,3-三氟丙烯的选择性之和为7.9%。
实施例12
与实施例1相同的操作,所不同的是将原料Z-1-氯-3,3,3-三氟丙烯替换为等物质的量的E-1-氯-3,3,3-三氟丙烯。反应结果如下:E-1-氯-3,3,3-三氟丙烯的转化率为22.3%,3,3,3-三氟丙炔的选择性为96.1%,Z-1-氯-3,3,3-三氟丙烯和2-氯-3,3,3-三氟丙烯的选择性之和为3.9%。
实施例13
与实施例1相同的操作,所不同的是将原料Z-1-氯-3,3,3-三氟丙烯替换为等物质的量的2-氯-3,3,3-三氟丙烯。反应结果如下:2-氯-3,3,3-三氟丙烯的转化率为25.7%,3,3,3-三氟丙炔的选择性为96.2%,E-1-氯-3,3,3-三氟丙烯和Z-1-氯-3,3,3-三氟丙烯的选择性之和为3.8%。
实施例14
与实施例1相同的操作,所不同的是将原料Z-1-氯-3,3,3-三氟丙烯替换为等物质的量的E-1,3,3,3-四氟丙烯。反应结果如下:E-1,3,3,3-四氟丙烯的转化率为46.8%,3,3,3-三氟丙炔的选择性为30.1%,Z-1,3,3,3-四氟丙烯和2,3,3,3-四氟丙烯的选择性之和为69.8%。
- 气相脱卤化氢制备3,3,3-三氟丙炔的方法
- 气相脱卤化氢制备3,3,3-三氟丙炔的方法