识别血液中基因异常的方法及系统
文献发布时间:2023-06-19 11:39:06
本申请请求了2018年9月3日提交的美国临时专利申请第62/726,337号的优先权的权益,其全部内容通过引用并入本说明书中。
本发明在一些实施例中涉及生物信息学,并且更具体地但不限于涉及一种用于识别母体血液中的基因疾病的方法及系统。
决定一胎儿中的多个遗传序列的拷贝数目具有重要的诊断价值。例如:在一显性遗传性疾病中,引起疾病的一等位基因的一单一拷贝的存在导致所述遗传性疾病的表现型的表达。相反,在一隐性遗传性疾病中,引起疾病的等位基因的一单一拷贝的存在仅导致所述个体成为携带者,而不引起所述遗传性疾病的所述表现型的表达。另外,多个遗传序列的多个异常的拷贝数,例如:部分或完全非整倍性的染色体片段或整个染色体,经常引起各种遗传性疾病。例如:三染色体症21会导致唐氏综合症(DS)。
目前,使用常规的细胞遗传学分析(例如:核型分析)或DNA分析来执行产前诊断,所述多个常规的细胞遗传学分析需要通过羊膜穿刺术、绒毛膜绒毛取样或脐带穿刺术获得胎儿遗传物质。然而,这些是多个侵入性的方法,并且与例如胎儿丢失的一风险等多个风险相关联。
允许在相对较短的时间内对整个基因组进行测序的多个技术的出现以及所述孕妇的血液中循环的细胞游离DNA(cfDNA)包括母源性及胎儿DNA的发现为非侵入式分析胎儿遗传物质提供了机会,而没有与多个侵入性采样方法相关的多个风险。本领域已知多个非侵入性产前诊断技术,用于识别多个特定的染色体异常以及识别起源于父系的多个单基因疾病。还已知多个非侵入性产前诊断技术用于排除起源于母系的多个单基因疾病、胎儿性别决定及胎儿恒河猴D基因分型。
发明内容
根据本发明的一些实施例,本发明提供了一种胎儿基因分型的方法,所述方法包括以下步骤:接收与一胎儿成对配对的母源性基因组DNA(genomic DNA,gDNA)数据、母源性细胞游离DNA(cell-free DNA,cfDNA)数据以及父源性基因组DNA数据;分析所述数据,以识别出一第一套的多个位点以及一第二套的多个位点,在所述第一套的多个位点,所述父母的多个不同的等位基因是纯合子的,在所述第二套的多个位点,所述父母中的至少一者具有一突变;对于所述第一套的每个位点,决定所述母源性细胞游离DNA数据的一相应部分是从所述胎儿中获得的一概率;及根据所述多个概率将所述第二套的每个位点分类为胎儿或母体,以对所述胎儿进行基因分型。
根据本发明的一些实施例,所述分析的步骤包括:在覆盖所述第一套的多个位点的多个读段中,识别一第一组的多个读段及一第二组的多个读段,所述第一组的多个读段仅包括表示多个父源性等位基因的多个读段,但不包括其他读段,所述第二组的多个读段包括所有其他的读段,并且其中所述决定所述概率的步骤是基于所述第一组中的多个读段与所述第二组中的多个读段之间的多个差异。
根据本发明的一些实施例,所述决定所述概率的步骤是基于至少一个序列比对图谱(sequence alignment map,SAM)参数。
根据本发明的一些实施例,所述序列比对图谱参数是选自于由观察的模板长度、与长度相关的胎儿分数、紧密异质间隙比对报告字串、配对段的CIGAR字符串、核苷酸序列、配对段的核苷酸序列、指示是否为一成对的读段的读段比对标记、指示是否一读段映射正确成对的读段比对标记、指示是否为一未映射的读段的读段比对标记、指示是否为一未映射的配对段的读段比对标记、指示是否为所述反向链上的一读段的读段比对标记、指示是否为在所述反向链的一配对段的读段比对标记、指示是否为第一个成对的一读段的读段比对标记、指示是否为第二个成对的一读段的读段比对标记、指示是否为非一主要比对的一读段的读段比对标记、指示是否为一平台及/或一供应商质量检查不及格的一读段的读段比对标记、指示是否为一PCR或光学重复的一读段的读段比对标记、指示是否为一补充比对的一读段、配对段的标记、映射质量、配对段的映射质量、染色体的多个基因组坐标、染色体上绝对起始位置的多个基因组坐标、染色体上绝对末端位置的多个基因组坐标、按染色体长度标准化的起始位置的多个基因组坐标、按染色体长度标准化的末端位置的多个基因组坐标、配对段的多个基因组坐标、G及C核苷酸的数目除以读段长度、多个G及C核苷酸的数目除以配对段的读段序列中的读段长度、在所述读段的一核苷酸序列中的A及/或C及/或G及/或T核苷酸的比率、在所述配对段的所述核苷酸序列中的A及/或C及/或G及/或T核苷酸的比率、有关所述读段或所述读段的配对段起源的一变异的信息、包括表A.1中出现的至少一特征、所述核苷酸序列中的Kmer的组成、所述配偶的核苷酸序列中的多个Kmer的组成、核苷酸质量序列、配对段的核苷酸质量序列、核苷酸质量序列的平均值及/或标准差及/或中位数、核苷酸质量序列的平均值及/或标准差及/或中位数、所述核苷酸质量序列中的Kmer组成、所述配对段的核苷酸质量序列中的Kmer组成、多个甲基化核苷酸的数目除以读取长度以及多个特定位置的甲基化所组成的群组。
根据本发明的一些实施例,所述决定所述概率的步骤至少是基于一观察的模板长度。
根据本发明的一些实施例,所述决定所述概率的步骤至少是基于与一长度有关的胎儿分数。
根据本发明的一些实施例,所述决定所述概率的步骤至少是基于一紧密异质间隙比对报告字符串。
根据本发明的一些实施例,所述决定所述概率的步骤至少是基于一核苷酸序列。
根据本发明的一些实施例,所述决定所述概率的步骤至少是基于配对段的核苷酸序列。
根据本发明的一些实施例,所述决定可能基于至少一读段比对标记,所述读段比对标记是选自于由指示是否为一成对的读段的一读段比对标记、指示是否一读段映射正确成对的读段比对标记、指示是否为一未映射的读段的一读段比对标记、指示是否为一未映射的配对段的一读段比对标记、指示是否为所述反向链上的一读段的一读段比对标记、指示是否为在所述反向链的一配对段的一读段比对标记、指示是否为第一个成对的一读段的读段比对标记、指示是否为第二个成对的一读段的一读段比对标记、指示是否为非一主要比对的一读段的一读段比对标记、指示是否为一平台及/或一供应商质量检查不及格的一读段的一读段比对标记、指示是否为一PCR或光学重复的一读段的一读段比对标记、指示是否为一补充比对的一读段所组成的群组。
根据本发明的一些实施例,所述方法还包括计算一总胎儿分数的步骤,其中所述分类的步骤还包括使用所述总胎儿分数。
根据本发明的一些实施例,所述方法还包括计算一总胎儿分数,并构建一胎儿大小分布及一母体大小分布,其中所述分类的步骤包括对所述胎儿大小分布进行分箱,并且针对每一片段大小箱计算一胎儿分数,以及基于所述片段所属的一对应的片段大小箱的一胎儿分数,对于至少一个位点及在所述至少一个位点处的至少一个片段计算所述片段是胎儿的一概率。
根据本发明的一些实施例,所述分类的步骤包括应用一贝叶斯方法。
根据本发明的一些实施例,所述贝叶斯方法包括使用至少一个所述父母的测序数据来计算的多个先验概率。
根据本发明的一些实施例,所述测序数据包括全基因组测序(whole genomesequencing,WGS)。
根据本发明的一些实施例,所述测序数据包括全外显子组测序(whole exomesequencing,WES)。
根据本发明的一些实施例,所述方法还包括使用机器学习来重新校准所述贝叶斯方法的输出。
根据本发明的一些实施例,所述分类的步骤包括将一机器学习程序应用于所述多个识别出的位点,以对所述胎儿进行基因分型。
根据本发明的一些实施例,所述机器学习程序包括深度学习程序。
根据本发明的一些实施例,所述方法还包括重新校准所述机器学习的输出。
根据本发明的一些实施例的一方面,提供了一种胎儿基因分型的方法,所述方法包括以下步骤:接收与一胎儿成对配对的母源性基因组DNA(gDNA)数据、母源性细胞游离DNA数据(cfDNA)以及父源性基因组DNA数据访问存储了一机器深度学习程序的一计算机可读介质,所述机器深度学习程序被训练用于将所述数据中的多个位点分类为胎儿或母亲;向所述方法提供所述数据;及从所述程序接收指示所述数据中的多个胎儿位点的一输出,从而对所述胎儿进行基因分型。
根据本发明的一些实施例,所述方法应用于所述母源性细胞游离DNA数据的多个杂合位点。
根据本发明的一些实施例,所述方法应用于所述母源性细胞游离DNA数据的多个纯合位点。
根据本发明的一些实施例,所述基因分型的步骤包括识别起源于父系的胎儿单基因疾病(single-gene disorder,SGD)。
根据本发明的一些实施例,所述基因分型的步骤包括识别起源于母系的胎儿单基因疾病(SGD)。
根据本发明的一些实施例,所述基因分型的步骤包括预测遗传的多个插入与多个缺失。
根据本发明的一些实施例,所述基因分型的步骤包括识别一胎儿染色体异常。
根据本发明的一些实施例,所述方法应用于执行一单基因疾病的非侵入性产前诊断(NIPD)。
根据本发明的一些实施例,所述方法应用于执行一多基因疾病的非侵入性产前诊断(NIPD)。
根据本发明的一些实施例,所述方法应用于执行一遗传性疾病的非侵入性产前诊断(NIPD),所述遗传性疾病选自于由布鲁姆综合征(Bloom Syndrome)、卡纳万病(CanavanDisease)、囊性纤维化(Cystic fibrosis)、家族性自主神经异常(FamilialDysautonomi)、莱利-戴综合征(Riley-Day syndrome)、范科尼贫血(C组)(Fanconi Anemia(Group C))、高雪氏病(Gaucher Disease)、第1a型糖原贮积病(Glycogen storagedisease 1a)、枫糖尿病(Maple syrup urine disease)、第IV型粘脂质贮积病(Mucolipidosis IV)、尼曼-匹克氏病(Niemann-Pick Disease)、泰-萨综合征(Tay-Sachsdisease)、β地中海贫血症(Beta thalessemia)、镰状细胞性贫血(Sickle cell anemia)、阿尔法地中海贫血症(Alpha thalessemia)、β地中海贫血症(Beta thalessemia)、第XI因子缺乏症(Factor XI Deficiency)、弗里德里希共济失调(Friedreich's Ataxia)、脂肪酸代谢障碍(MCAD)、帕金森疾病-青少年(Parkinson disease-juvenile)、连接蛋白26(Connexin26)、脊髓性肌萎缩(SMA)、雷特综合征(Rett syndrome)、苯丙酮尿症(Phenylketonuria)、贝克尔肌营养不良症(Becker Muscular Dystrophy)、杜兴氏肌营养不良症(Duchennes Muscular Dystrophy)、脆性X综合征(Fragile Xsyndrome)、A型血友病(Hemophilia A)、早发性阿尔茨海默病(Alzheimer dementia-early onset)、乳腺癌/卵巢癌(Breast/Ovarian cancer)、结肠癌(Colon cancer)、糖尿病/青少年发病的成人型糖尿病(MODY)、亨廷顿病(Huntington disease)、强直性肌肉营养不良(Myotonic MuscularDystrophy)、早发性帕金森病(Parkinson Disease-early onset)、皮兹-杰格斯综合症(Peutz-Jeghers syndrome)、多囊肾病(Polycystic Kidney Disease)、扭转性肌张力障碍(Torsion Dystonia)所组成的群组。
根据本发明的一些实施例,所述方法应用于执行用于一遗传性疾病的非侵入性产前诊断(NIPD),并且所述方法还包括对所述遗传性疾病进行产前或产后治疗。
根据本发明的一些实施例,所述方法应用于执行用于一先天性疾病的非侵入性产前诊断(NIPD)。
根据本发明的一些实施例,所述先天性疾病选自于畸形、神经管缺陷、染色体异常、唐氏综合症(Down syndrome)(或21三体症(trisomy 21))、18三体症(Trisomy 18)、脊柱裂(spina bifida)、唇腭裂(cleft palate)、泰-萨病(Tay Sachs disease)、镰状细胞性贫血(sickle cell anemia)、地中海贫血(thalassemia)、囊性纤维化(cystic fibrosis)、亨廷顿氏病(Huntington's disease)及脆性x综合征(fragile x syndrome)所组合的群组。
根据本发明的一些实施例,所述染色体异常选自于由唐氏综合症(额外的21号染色体)、特纳综合征(Turner Syndrome)(45X0)及克氏综合征(Klinefelter'ssyndrome)(一男性具有2个X染色体)。
根据本发明的一些实施例,所述畸形包括一肢体畸形。
根据本发明的一些实施例,所述肢体畸形是选自于由缺肢症(amelia)、缺指症(ectrodactyly)、短肢症(phocomelia)、多肢症(polymelia)、多指性(polydactyly)、并指症(syndactyly)、多并趾症(polysyndactyly)、少指症(oligodactyly)、短指症(brachydactyly)、软骨发育不全(achondroplasia)、先天性发育异常(congenitalaplasia)或发育不全(hypoplasia)、羊膜带综合征(amniotic band syndrome)及锁骨颅骨发育异常症(cleidocranial dysostosis)所组成的群组。
根据本发明的一些实施例,所述畸形包括一心脏的先天性畸形。
根据本发明的一些实施例,所述先天性畸形选自于由动脉导管未闭(patentductus arteriosus)、房中隔缺损(atrial septal defect)、室中隔缺损(ventricularseptal defect)及法洛四联症(tetralogy of fallot)所组成的群组。
根据本发明的一些实施例,所述畸形包括所述神经系统的一先天性畸形。
根据本发明的一些实施例,其中所述神经系统的所述先天性畸形是选自于由多个神经管缺损(例如:脊柱裂(spina bifida)、脑膜膨出(meningocele)、脑膜脊髓膨出(meningomyelocele)、脑膨出(encephalocele)及无脑畸形(anencephaly))、爱纳尔德一查理综合征(Arnold-Chiari malformation)、丹迪-沃克畸形(Dandy-Walkermalformation)、脑积水(hydrocephalus)、小头症(microencephaly)、大头症(megencephaly)、平脑症(lissencephaly)、多小脑回症(polymicrogyria)、全前脑症(holoprosencephaly)及胼胝体发育不全(agenesis of the corpus callosum)所组成的群组。
根据本发明的一些实施例,所述畸形包括所述胃肠系统的先天性畸形。
根据本发明的一些实施例,所述胃肠系统的所述先天性畸形是选自由狭窄(stenosis)、闭锁(atresia)及肛门闭锁(imperforate anus)所组成的群组。
根据本发明的一些实施例,所述方法还包括对所述先天性疾病进行产前或产后治疗。
根据本发明的一些实施例,所述方法还包括对所述畸形施行产前或产后治疗。
根据本发明的一些实施例,所述方法还包括施行产前或产后治疗是选自于由基于药物的干预、手术、基因治疗、营养治疗及其组合所组成的群组。
根据本发明的一些实施例,所述方法还包括执行一妊娠终止。
根据本发明的一些实施例,所述方法还包括当所述基因分型指示胎儿异常或基因异常时取得胎儿遗传物质,以及分析所述胎儿遗传物质,以决定至少所述胎儿是否具有所述异常或基因异常。
根据本发明的一些实施例,提供了一种计算机软件产品,所述计算机软件产品包括一计算机可读介质,所述计算机可读介质中存储多个程序指令,所述多个指令在被一数据处理器读取时使一数据处理器接收与一胎儿成对配对的母源性细胞游离DNA数据及父源性细胞游离DNA数据,以执行如上面描述的方法,并且优选地如下面进一步详细描述。
根据本发明的一些实施例,提供了一种胎儿基因分型系统,所述胎儿基因分型系统包括:一输入电路,接收与一胎儿成对配对的母源性细胞游离DNA(cfDNA)数据及父源性细胞游离DNA(cfDNA)数据;及一数据处理器,被配置为用于分析所述数据来识别所述父母为纯合子的多个位点,以执行如上面描述的方法,并且优选地如下面进一步详细描述。
除非另外定义,否则本文所使用的所有技术术语和/或科学术语都具有与本发明所属领域的普通技术人员通常所理解的相同的意义。虽然本发明的实施方式可以通过类似或等同于本发明的实施方式所述的任何方法和材料实施或测试,本发明的实施方式、列举的方法和/或材料已在下面描述。在冲突的情况下,将以本专利说明书并且包括定义为准。此外,材料、方法和实施方式仅是举例性质,并且不必然用以限制。
本发明的方法及/或系统的实施涉及手动地、自动地或其组合来执行或完成所选定的多个任务。此外,根据本发明的方法及/或系统的多个实施例的实际仪器和装置,可以使用一操作系统,通过硬件、软件、或固件或其组合来实施若干所选定的步骤。
例如,根据本发明实施例的用于执行所选任务的硬件可以实施为一芯片或一电路。作为软件,本发明的所选定的任务可以实施为计算机使用的任何合适的操作系统所执行的多个软件指令。在本发明的一示例性实施例中,根据本发明的方法及/或系统的多个选定的一个或多个任务可以被描述为通过一数据处理器执行,例如:一种用于执行多个指令的计算平台。可选地,所述数据处理器包括用于存储多个指令及/或数据的一易失性存储器,及/或一非易失性存储器,例如:一磁性硬盘及/或可移动介质。可选地,本发明还提供了一网络连接,本发明还可选地提供了一显示器及/或一用户输入设备,例如:一键盘或鼠标。
附图说明
本发明的一些实施例在此仅通过举例的方式并参考多个附图来描述,通过详细说明附图具体的参考资料,应当强调所示的细节仅为举例,用以说明本发明实施例的目的。基于这点,结合所述附图及描述使得本领域技术人员能清楚的实施本发明的实施例。
在附图中:
图1是根据本发明的一些实施例的用于一非侵入性产前变异侦测的一流水线的一示意图。
图2A至2D示出了根据本发明的一些实施例进行的多个实验中获得的准确度及多个后验概率之间的关系,图2A至2C示出了由亲本基因型决定的三类单核苷酸多态性(SNP)位置中的G1至G4家族的多个结果及G1的子采样数据(“多个模拟”)。所述x轴显示所述多个预测的后验概率的所述最小阈值,所述阈值指示所包含的基因座的决定性水平,所述y轴指示所述后验概率高于所述阈值的所有位点之间的准确度。所述总准确度出现在所述x轴的最低点,在所述地方设置了最大后验概率的最低可能阈值(在图2A及2B中,每一基因座有两种可能的胎儿基因型,所述数值为0.5;并且在图2C中,所述数值为0.33,多个因为所有3种胎儿基因型都是可能的),还示出了在每一阈值处计算所述准确度的总基因座的多个计数。
图3A至3D示出了在一母亲是杂合时的多个插入/缺失位点的校准,如同在根据本发明的一些实施例进行的多个实验中所获得的。图3A至3D的所述格式类似于图2A至2D,但用于多个插入缺失。多个基因座之间的多个准确度与高于出现在所述x轴上的所述多个阈值的一后验概率一同示出。图3A至C3中显示了相同的三类基因组位点,每一类别的总准确度是所述x轴上最左点的所述准确度(图3A及3B中为0.5,图3C中为0.3)。每一子图的所述底部显示了每一阈值处用于准确度计算的基因座的所述计数。
图4A至4C示出了如根据本发明的一些实施例进行的多个实验中取得的如图2A至3D所示的所述三类基因座处的作为一测序深度及一胎儿分数的一函数的准确度的多个热图。
图5A至5G示出了如在根据本发明的一些实施例进行的多个实验中取得的多个在妊娠前三个月的个案的相对多个SNP及多个插入缺失的性能。多个基因座的多个准确度与高于出现在所述x轴上的所述多个阈值的一后验概率一同示出。根据所述先前使用的多个基因座类别,给出了针对多个SNP(图5A至5C)及多个插入缺失(图D至F)的所述多个实验中测序的多个家族的多个发现,每一类别的总准确度是所述x轴上最左点的所述准确度(A至B及D至E中为0.5,C及F中为0.3),还示出了从每一阈值的所述准确度中计算出的所述多个总计数。
图6A至6F示出了如在根据本发明的一些实施例执行的多个实验中获得的变异概率重新校准的接收者操作特征(ROC)曲线,基于一机器学习的变异重新校准步骤之前及之后,对于多个相同的先前使用的基因座类别示出了所述两个测试套组的多个ROC曲线。对于每一曲线,还显示了所述曲线下的所述微平均面积(AUC)及所述总准确度(ACC)。
图7A至7G示出了如在根据本发明的一些实施例进行的多个实验中获得的所述准确度的校准,示出了在变量重新校准前后的所述准确度作为所述预测概率的一函数。所述“完美校准”线表示被分配给一预测的所述概率完美地描述了正确的所述概率(就准确度而言)的一情况,还示出了每一分箱中基因座的边缘分布。
图8A至8G展示了执行基于机器学习的变异重新校准来过滤多个变异的能力,多个基因座的多个准确度与高于出现在所述x轴上的所述多个阈值的一后验概率一同示出。在变异重新校准前后皆会提供多个发现。每一类别的总准确度是所述x轴上的最左点的所述准确度,还示出了从每一阈值的所述准确度中计算出的所述多个总计数。
图9A至9G显示如在根据本发明的一些实施例进行的多个实验中获得的多个细胞游离DNA片段长度分布,列出了所述多个实验中包括的多个家庭的多个胎儿(蓝色)及母源性(绿色)片段的长度分布。
图10示出了如在根据本发明的一些实施例进行的多个实验中获得的多个突变验证桑格(Sanger)结果,使用桑格确认了多个突变家族E1及G5。第一行示出了与所述参考等位基因(G)纯合的一对照组,第二行示出了所述父亲,所述父亲是所述变异的等位基因(T)的一携带者,第三行及第四行示出了所述胎儿的所述多个结果,一次使用一正向引物,一次使用一反向引物,所述胎儿与所述变异的等位基因是纯合的。
如图11是总结了一贝叶斯模型(在图11中称为霍巴里(Hoobari))及端到端深度学习模型(在图11中称为深度霍巴里)之间的所述多个差异的一图示。
图12A至12P示出了根据本发明的一些实施例使用一机器深度学习程序执行的一实验的一第一阶段的多个结果,示出了使用四个经过测试的网络对子采样数据集G1中的多个SNP进行的准确度及损失结果。
图13A至13P示出了根据本发明的一些实施例使用一机器深度学习程序执行的一实验的多个独热(one-hot)及群热(group-hot)编码结果。
图14A至14P示出了当仅使用父母信息中的多个基因型时,根据本发明的一些实施例使用一机器深度学习程序进行的一实验的一第一阶段的多个结果。
图15是根据本发明的各个示例性实施例的适合于胎儿基因分型的一方法的一流程图。
图16是根据本发明的一些实施例的可用于胎儿基因分型的一服务器-客户端计算机配置的一示意图。
具体实施方式
本发明在一些实施例中涉及生物信息学,并且更具体地但不限于涉及一种用于识别母体血液中的基因疾病的方法及系统。
在详细解释本发明的至少一个实施方式之前,应当理解本发明不一定限定于本发明的构造的细节,以及在以下描述和/或附图图示/或实施方式所阐述的组件和/或方法的排列,本发明能够以其他实施方式或以各种方法来实施或应用。
图15是根据本发明的各个示例性实施例的适合于胎儿基因分型的一方法的一流程图。应当理解,除非另外定义,否则以下描述的多个操作可以同时或顺序地以许多组合或执行顺序来执行。具体地,所述多个流程图的所述顺序不应认为是限制性的。例如:以下描述或所述多个流程图中以一特定的顺序出现的两个或更多个操作可以以一不同的顺序(例如:一相反的顺序)或实质上同时执行。此外,以下描述的几种操作是可选的及可能不执行。
本多个实施例的多个处理操作可以以多种形式实施,例如:所述多个处理操作可以实施在一有形的介质中,例如:用于执行所述多个操作的一计算机,所述多个处理操作可以实施在一计算机可读介质上,所述计算机可读介质包括多个用于执行所述多个方法操作的多个计算机可读指令,所述多个处理操作也可以实施在具有多个数字计算机功能的电子设备中,所述数字计算机功能被安排为在所述有形介质上运行所述计算机程序或在一计算机可读介质上执行所述指令。
实施根据本发明的一些实施例的所述方法的多个计算机程序通常可以在一分配介质上分配给多个用户,所述分配介质例如但不限于:CD-ROM、多个闪存设备、多个闪存驱动器,或者在一些实施例中,可通过互联网(例如:在一云端环境中)或通过一蜂窝网络进行网络通信的方式访问的多个驱动器。可以将所述多个计算机程序从所述分配介质复制到一硬盘或一类似的中间存储介质。可以通过将所述多个计算机指令从所述多个计算机指令的分布介质或所述多个计算机指令的中间存储介质中加载到所述计算机的所述执行存储器中,并且配置所述计算机使所述计算机根据本发明的所述方法运作来运行所述多个计算机程序。实现根据本发明一些实施例的所述方法的多个计算机程序也可以由属于一云计算环境的一个或多个数据处理器执行。所有所述多个操作对于计算机系统领域的多个技术人员来说都是已知的。由本多个实施例的所述方法使用及/或提供的数据可以借助于网络通信通过所述互联网、通过蜂窝网络或通过任何适合于数据传输的网络类型来传输。
根据本发明的多个优选实施例的所述方法可以被嵌入到多个医疗保健系统中,并且可以允许识别多个胎儿异常或疾病,例如但不限于:起源于父亲及/或母亲的多个胎儿单基因疾病(SGDs)、多个遗传性插入缺失、多个染色体异常及/或多个单基因或多基因疾病,包括但不限于:布鲁姆综合征、卡纳万病、囊性纤维化、家族性自主神经异常、莱利-戴综合征、范科尼贫血(C组)、高雪氏病、第1a型糖原贮积病、枫糖尿病、第IV型粘脂质贮积病、尼曼-匹克氏病、泰-萨综合征、β地中海贫血症、镰状细胞性贫血、阿尔法地中海贫血症、β地中海贫血症、第XI因子缺乏症、弗里德里希共济失调、脂肪酸代谢障碍(MCAD)、帕金森疾病-青少年、连接蛋白26、脊髓性肌萎缩(SMA)、雷特综合征、苯丙酮尿症、贝克尔肌营养不良症、杜兴氏肌营养不良症、脆性X综合征、A型血友病、早发性阿尔茨海默病、乳腺癌/卵巢癌、结肠癌、糖尿病/青少年发病的成人型糖尿病(MODY)、亨廷顿病、强直性肌肉营养不良、早发性帕金森病、皮兹-杰格斯综合症、多囊肾病及扭转性肌张力障碍。
根据本发明的多个优选实施例的所述方法可以允许识别一胎儿先天性疾病,例如:神经管缺陷、染色体异常、唐氏综合症(或21三体症)、18三体症、脊柱裂、唇腭裂、泰-萨病、镰状细胞性贫血、地中海贫血、囊性纤维化、亨廷顿氏病、脆性x综合征、特纳综合征(45X0)及克氏综合征(一男性具有2个X染色体)。
根据本发明的多个优选实施例的所述方法可以允许识别一畸形,例如但不限于:一肢体畸形,例如:缺肢症、缺指症、短肢症、多肢症、多指性、并指症、多并趾症、少指症、短指症、软骨发育不全、先天性发育异常或发育不全、羊膜带综合征及锁骨颅骨发育异常症;所述心脏的一先天性畸形,例如:动脉导管未闭、房中隔缺损、室中隔缺损及法洛四联症;所述神经系统的一先天性畸形,例如:多个神经管缺损(例如:脊柱裂、脑膜膨出、脑膜脊髓膨出、脑膨出及无脑畸形)、爱纳尔德一查理综合征、丹迪-沃克畸形、脑积水、小头症、大头症、平脑症、多小脑回症、全前脑症及胼胝体发育不全;所述胃肠系统的一先天性畸形,例如:狭窄、闭锁及肛门闭锁。
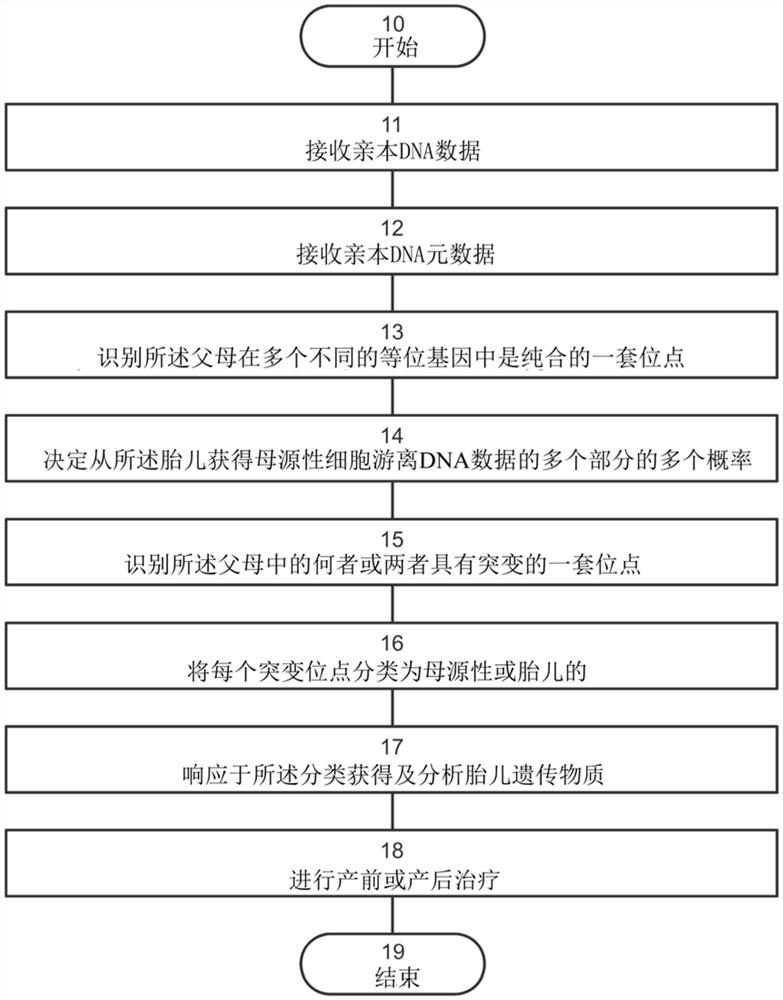
所述方法从10开始并继续到11,在11处接收与一胎儿有关的一对DNA数据,可以通过所述方法从存储所述DNA数据的一计算机可读介质中接收所述DNA数据,所述DNA数据优选至少包括母源性基因组DNA(gDNA)数据、母源性细胞游离DNA(cfDNA)数据及父源性基因组DNA数据。在本发明的一些实施例中,所述DNA数据仅包括多个基因型,并且没有读段水平的信息,或者没有核苷酸水平的信息、或者皆没有读段水平的信息及核苷酸水平的信息。
如本说明书所用的“基因型”是指在一给定的父母或胎儿细胞中,在一给定的遗传基因座或一套相关的基因座处的多个等位基因的所述二倍体组合。一纯合的对象携带两个拷贝的相同等位基因,而一杂合的对象携带两个不同的等位基因。在具有两个等位基因“A”及“a”的一基因座的最简单情况下,可以形成三种基因型:A/A、A/a及a/a,可能存在两种以上的等位基因的形式,因此,可能存在三种以上的基因型。
所述父母的所述多个基因型可以通过基因分型来决定。
如本说明书所用,对一对象(或DNA样品)的一(多个)基因的一多态性等位基因进行“基因分型”是指检测在一对象(或一样品)中存在的所述(多个)基因为何种(多个)等位基因或多态性形式。
如本说明书所用,术语“基因”或“重组基因”是指一核酸分子包括一开放阅读框,并且包含至少一外显子及一(可选地)内含子序列。术语“内含子”是指存在于一给定基因中的一DNA序列,所述DNA序列在mRNA成熟过程中被剪接。
可选地,所述方法继续到12,在12处接收到与一个或两个父母有关的元数据,所述接收的元数据可选地及优选地包括附件1的表A.1中列出的所述多个特征中的至少一个,更优选为多于一个。
所述方法继续进行到13,在13中对所述数据进行分析,以识别所述父母在多个不同的等位基因中是纯合的第一套位点。可选地并且优选地至少对所述母源性DNA数据的每一位点执行所述操作。可以使用一变异侦测技术在多个个体中执行操作13,所述操作基于DNA的测序与所述基因组的比对,应用一商业上取得的变异侦测器,以及从所述得到的变异列表中筛选或选择所述父母在多个不同的等位基因为纯合的多个所述多个位点。
可以根据本发明的一些实施例使用的多个序列比对技术包括但不限于:伯罗斯惠勒比对器(BWA)、ABA、ALE、AMAP、阿农(anon)、BAli-Phy、碱基到碱基、BHAOS/DIALIGN、领结(Bowtie)、领结2(Bowtie 2)、ClustalW、密码子代码(CodonCode)比对器、科马斯(Comass)、DECIPHER、DIALIGN-TX、DIALIGN-T、DNA比对、DNA碱基序列汇编程序、EDNA、FSA、遗传(Geneious)、Kalign、MAFFT、MARNA、MAVID、MSA、MSAProbs、MULTALIN、Multi-LAGEN、肌肉(MUSCLE,)、蛋白石(Opal)、山核桃(Pecan)、菲洛(Phylo)、普拉林(Praline)、PicXAA、POA、普罗巴林(Probalign)、ProbCons、PROMALS3D、PRRN/PRRP、PSAlign、RevTrans、SAGA、SAM、Se-Al、星星(STAR)、星星融合(STAR-Fusion)、StatAlign、Stemloc、T-咖啡(T-Coffee)、优基因(UGENE)、向量朋友(VectorFriends)及GLProbs
适用于本实施例的示例性变异侦测器包括但不限于:基因组分析工具包(GATK)及免费贝叶斯(Freebayes)。例如:Freebayes可包括基于与一特定靶标比对的多个读段的多个文字序列的一比对,而不是其精确比对。GATK可以包括:(i)预处理;(ii)变异发现,及(iii)侦测集改进。预处理可以包括从原始序列数据(例如:FASTQ或uBAM格式)开始,并且生成多个分析就绪的BAM文件,处理过程可以包括与一参考基因组的比对以及多个数据清理操作,以纠正多个技术偏见,并且制作适用于分析的所述数据,变异发现可以包括从多个分析就绪的BAM文件开始,并且以VCF格式生成一侦测集,处理过程可能涉及识别一个或多个个体显示可能的基因组变异的多个位点,并且应用适合于实验设计的多个过滤方法,侦测集改进可以包括以一VCF侦测集开始及结束,处理可以涉及使用元数据,以评估及提高基因分型的准确度,附加更多信息并且评估所述侦测集的整体质量。
还设想了多个变异侦测器,例如但不限于:鸭嘴兽(Platypus)、VarScan、领结(Bowtie)分析、MuTect及/或SAM工具包(SAMtools)。例如:领结(Bowtie)分析可以包括实现用于比对的伯罗斯惠勒(Burrows-Wheeler)转换。MuTect可以包括:(i)预处理;(ii)统计分析;及(iii)后处理。预处理可以包括多个测序读段的一初始比对,统计分析可以包括使用两个贝叶斯分类器及一分类器,所述分类器可以检测一SNP在一给定的位点是否为非参考,对于发现为非参考的所述多个位点,则为其他分类器可确保为正常不携带SNP,后处理可包括去除测序伪影、短读段比对及杂交捕获。SAM工具包(SAMtools)可以包括存储、操作及比对被存储为多个SAM文件的多个测序读段。
在本发明的各个示例性实施例中,所述方法进行到14处,在所述14中,对于第一套的每一位点,决定从所述胎儿获得母源性细胞游离DNA数据的各个部分的一概率。操作14是有利的,由于操作14允许所述方法获得与来自于胎儿的读段及来自于母亲的读段之间的所述母源性DNA数据中的所述多个差异有关的信息,这可以通过在第一套中搜索呈现父源性等位基因的多个读段,并且将被一个或多个所述多个读段覆盖的每一位点以及优选地没有被其他读段所覆盖的每一位点定义为来自于胎儿的一位点来完成。所述第一套的所有来自于胎儿的位点在所述第一套内定义一第一组,所述第一套中的多个其余位点在第一套中定义一第二组。覆盖所述第一组中的所述多个位点的所述多个读段的所述特征与覆盖所述第二组中的所述多个位点的所述多个读段的特征之间的所述多个差异随后被用于决定所述母源性细胞游离DNA数据中一特定位点是来源自胎儿的所述概率。
所述方法也可以进行到15,在所述15中分析所述数据,以识别一第二套位点,所述第二套位点中所述父母中的至少一个具有一突变。可以使用以上针对操作13所述的任何一种商业上可取得的技术来执行操作15,除了操作15以外,除了所述父母中的没有一个具有一突变的多个位点从所述输出中被过滤移除。所述第二套优选地包括不属于所述第一套的至少一位点,并且更优选地多个位点。然而,所述第一套及第二套位点不必一定是不相交的,因为对于父母在不同等位基因是纯合子的一位点原则上可以呈现一突变。然而,由于已经在14处决定了所述第一套的每一位点的所述概率,因此,本多个实施例设想了其中所述第一套及所述第二套为多个不相交的套组的一方案。
所述方法进行到16,根据在14处获得的所述多个概率,第二套的每一位点被分类为胎儿或母体,然后可以提取被分类为胎儿的每一位点的所述基因型,从而对所述胎儿进行基因分型。
在本发明的一些实施例中,所述方法进行到17,在所述17中取得并分析了胎儿遗传物质。优选地,当在16处的所述基因分型指示出胎儿异常或基因异常时执行17,在这种情况下,所述分析17旨在决定至少所述胎儿是否具有所述异常或基因异常。
在本发明的一些实施例中,所述方法进行到18,在18中给予产前或产后治疗。例如:当所述方法在17或18中识别出一胎儿遗传性疾病时,可以对所述被识别的胎儿遗传性疾病进行产前或产后治疗。当所述方法在17或18中识别出一先天性疾病时,可以对所述被识别的先天性疾病进行产前或产后治疗。当所述方法在17或18中时发现畸形时,可以对所述畸形进行产前或产后的治疗。根据本发明的一些实施例,设想其他产前或产后治疗,包括但不限于:基于药物的干预、手术、基因治疗、营养治疗及其组合。在本发明的一些实施例中,执行一妊娠终止。
所述方法在19结束。
在14中的多个概率的决定及所述分类16可以以一种以上的方式来完成。
在本发明的一些实施例中,在14中的决定是基于一个或多个序列比对图谱(SAM)参数。SAM是用于存储生物数据(例如但不限于:核苷酸序列)多个的一已知格式,并且定义了各种参数(在本说明书中称为多个SAM参数)。所述SAM格式的规格的一说明可以在www(dot)samtools(dot)github(dot)io/hts-specs/SAMv1(dot)pdf上找到,其内容通过引用合并于此。
适用于本多个实施例的多个SAM参数的多个代表性示例包括但不限于:观察的模板长度、与长度相关的胎儿分数、紧密异质间隙比对报告字串、配对段的CIGAR字符串、核苷酸序列、配对段的核苷酸序列、指示是否为一成对的读段的读段比对标记、指示是否一读段映射正确成对的读段比对标记、指示是否为一未映射的读段的读段比对标记、指示是否为一未映射的配对段的读段比对标记、指示是否为所述反向链上的一读段的读段比对标记、指示是否为在所述反向链的一配对段的读段比对标记、指示是否为第一个成对的一读段的读段比对标记、指示是否为第二个成对的一读段的读段比对标记、指示是否为非一主要比对的一读段的读段比对标记、指示是否为一平台及/或一供应商质量检查不及格的一读段的读段比对标记、指示是否为一PCR或光学重复的一读段的读段比对标记、指示是否为一补充比对的一读段、配对段的标记、映射质量、配对段的映射质量、染色体的多个基因组坐标、染色体上绝对起始位置的多个基因组坐标、染色体上绝对末端位置的多个基因组坐标、按染色体长度标准化的起始位置的多个基因组坐标、按染色体长度标准化的末端位置的多个基因组坐标、配对段的多个基因组坐标、G及C核苷酸的数目除以读段长度、多个G及C核苷酸的数目除以配对段的读段序列中的读段长度、在所述读段的一核苷酸序列中的A及/或C及/或G及/或T核苷酸的比率、在所述配对段的所述核苷酸序列中的A及/或C及/或G及/或T核苷酸的比率、有关所述读段或所述读段的配对段起源的一变异的信息、包括表A.1中出现的至少一特征、所述核苷酸序列中的Kmer的组成、所述配偶的核苷酸序列中的多个Kmer的组成、核苷酸质量序列、配对段的核苷酸质量序列、核苷酸质量序列的平均值及/或标准差及/或中位数、核苷酸质量序列的平均值及/或标准差及/或中位数、所述核苷酸质量序列中的Kmer组成、所述配对段的核苷酸质量序列中的Kmer组成、多个甲基化核苷酸的数目除以读取长度以及多个特定位置的甲基化。
在本发明的一些实施例中,至少基于一观察的模板长度来决定在14中的所述概率,在本发明的一些实施例中,至少基于与一长度有关的胎儿分数来决定在14中的所述概率。在本发明的一些实施例中,在14中的所述概率至少基于一CIGAR字符串来决定,在本发明的一些实施例中,在14中的所述概率至少基于一核苷酸序列来决定,在本发明的一些实施例中,在14中的所述概率为至少基于一配对段的核苷酸序列来决定。
在本发明的一些实施例中,至少基于至少一个读段比对标记来决定在14中的概率。适用于本实施例的多个读段比对标记的多个代表性示例包括但不限于:所述决定可能基于至少一读段比对标记,所述读段比对标记是选自于由指示是否为一成对的读段的一读段比对标记、指示是否以正确成对来映射的一读段的一读段比对标记、指示是否为一未映射的读段的一读段比对标记、指示是否为一未映射的配对段的一读段比对标记、指示是否为所述反向链上的一读段的一读段比对标记、指示是否为在多个反向链的一配对段的一读段比对标记、指示是否为第一个成对的一读段读对的比对标记、指示是否为第二个成对的一读段比对标记、指示是否为非一主要比对的一读段的一读段比对标记、指示是否为一平台及/或一供应商质量检查不及格的一读段的一读段比对标记、指示是否为一PCR或光学重复的一读段的一读段比对标记、指示是否为一补充比对的一读段。
本多个实施例还设想了计算一总胎儿分数。在所述多个实施例中,所述分类16可选地及优选地基于所述计算的总胎儿分数。还设想了其中构建一胎儿大小分布及一母亲大小分布的多个实施例。在所述多个实施例中,所述分类16可选地及优选地包括对所述胎儿大小分布进行分箱,针对每个片段大小箱计算一胎儿分数,以及基于所述片段所属的一相应的片段大小箱的一胎儿分数,针对至少一个位点及至少一个位点处的至少一个片段来计算所述片段是胎儿的一概率。在一些实施例中,可选地及优选地,在16中的所述分类包括应用一贝叶斯程序,所述贝叶斯程序可选地及优选地包括使用所述父母中的至少一个的测序数据(例如:基因组测序或整个外显子组测序)计算的多个先验概率。
在本发明的一些实施例中,在14中的所述多个概率以及可选地及优选地还包括在16中的所述分类的决定包括应用一机器学习程序。
如本说明书中所使用的术语“机器学习”是指作为计算机程序的一程序来实施,所述计算机程序被配置为从先前收集的数据中导出多个模式、规律或规则,以对未来数据产生一适当的响应,或以一些有意义的方式描述所述数据。
适用于本多个实施例的多个机器学习程序的多个代表性示例包括但不限于:聚集、关联规则算法、特征评估算法、子集选择算法、支持向量机、分类规则、成本敏感分类器、投票算法、堆叠算法、贝叶斯算法网络、决策树、神经网络、卷积神经网络、基于示例的算法、线性建模算法、k最近邻(KNN)分析、集成学习算法、概率模型、图形模型、逻辑回归方法(包括多项逻辑回归方法)、梯度上升方法、奇异值分解方法及主成分分析。
以下是适用于本实施例的一些机器学习程序的一概述。
多个支持多个向量机是基于统计学习理论的多个演算法,根据本发明一些实施例的支持向量机(SVM)可以用于多个分类目的及/或用于数值预测。用于分类的一支持向量机在本说明书中被称为“支持向量分类器”,用于数值预测的支持向量机在本说明书中被称为“支持向量回归”。
一SVM通常以一内核功能为特征,所述内核功能的所述选择决定所得到的SVM是否提供分类、回归或其他功能。通过应用所述内核函数,所述SVM将多个输入向量配对到高维特征空间,可以在所述高维特征空间中构建一决策超表面(也称为一分隔符)以提供分类、回归或其他决策功能。在最简单的情况下,所述表面是一超平面(也称为一线性分隔符),但是也设想了使用多个更复杂的分隔符,并且可以使用多个内核函数来应用,定义所述超曲面的所述数据点被称为多个支持向量。
所述支持向量分类器选择一分隔符,其中所述分隔符与多个最接近的数据点的所述距离应尽可能大,从而将与一给定的分类中的多个对象相关联的多个特征向量点与与在所述分类之外的多个对象相关联的多个特征向量点分开。对于支持向量回归,构建具有可接受误差的一半径的一高维管道,所述管道可以最大程度地减少所述数据集的所述误差,同时还可以最大化所述关联曲线或函数的所述平坦度。换句话说,所述管道是所述拟合曲线周围的一外壳,由最接近所述曲线或表面的多个数据点的一集合来定义。
一支持向量机的一优点是一旦决定了所述多个支持向量,就可以从计算中删除所述多个剩下的观察值,从而大大降低了所述问题的所述计算复杂度。一SVM通常分为两个阶段:一训练阶段及一测试阶段。在所述训练阶段期间,将生成一组支持向量,以用于执行所述决策规则。在所述测试阶段,将使用所述决策规则来决定多个决策。一支持向量演算法是一种训练一SVM的一方法。通过执行所述演算法,将生成一组训练参数,包括表征所述SVM的所述多个支持向量。适用于本多个实施例的一支持向量演算法的一代表性示例包括但不限于顺序最小优化。
在KNN分析中,决定多个对象的所述亲和力或紧密度,所述亲和力也称为多个对象之间的一特征空间中的距离。基于所述多个决定的距离,将所述对象集合成簇并检测一离群值。因此,所述KNN分析是一种基于一对象距离所述特征空间中的第k个最近邻居的所述距离来寻找多个基于距离的离群值的一技术。具体而言,根据每个对象到所述对象的第k个最近邻居的距离对每个对象进行排名,最远的对象被宣告为多个离群值。在一些情况下,多个最远的对象被宣告为多个离群值。也就是说,例如:k个邻居及一特定距离,如果在等于或小于所述对象的所述特定距离处具有不大于k个对象,则一对象相对于多个参数是一离群值。所述KNN分析是使用监督学习一分类技术,呈现出一个项目并将所述项目与具有两个或多个分类的一训练集进行比较,所述项目被分配到所述项目的多个k最近邻居中最常见的所述类别。也就是说,计算到所述训练集中的所有项目的所述距离,以尋找所述最近的k,然后从所述k中提取多数分类,并且分配给项目。
关联规则算法是一种用于提取多个特征之间的多个有意义的关联模式的一技术。
在机器学习的上下文中,术语“关联”是指多个特征之间的任何相互关系,而不仅仅是预测一特定的分类或数值的多个特征之间的相互关系。关联包括但不限于尋找多个关联规则、多个尋找模式、执行特征评估、执行特征子集选择、开发多个预测模型以及理解多个特征之间的交互作用。
术语“关联规则”是指在所述多个数据集中频繁出现的多个元素,关联规则包括但不限于多个关联模式、多个区分模式、多个频繁模式、多个封闭模式及多个巨大模式。
关联规则演算法的一通常的主要步骤是找到所有观察值中最频繁出现的一组项目或特征,一旦获得所述列表,就可以从所述列表中提取多个规则。
前面提到的自组织图谱是一无监督的学习技术,通常用于对高维数据进行可视化及分析。多个典型的应用程序集中在所述图谱上的所述数据中的所述可视化的多个中心依赖关系上。所述演算法生成的所述图谱可用于加快其他演算法对多个关联规则的所述识别,所述演算法通常包括多个处理单元的一网格,称为“多个神经元”。每个神经元都与称为观察的一特征向量相关联。所述图谱尝试使用一组受限制的模型以最佳的准确度表示所有可用的观察值。同时,所述多个模型在所述网格上变得有序,以使多个相似的模型彼此靠近,而多个相异的模型彼此远离,所述程序允许所述数据中的所述多个要素之间的多个依赖关系或多个关联的所述识别及可视化。
多个特征评估算演算法是针对多个特征的所述排名或针对在基于所述多个特征的影响而对所述多个特征进行选择后的所述排名。
信息获取是适用于特征评估的所述多个机器学习方法中的一个,信息增益的定义需要熵的定义,所述熵是对训练多个示例的一集合中杂质的一度量。通过知道一特定特征的多个数值而发生的所述目标特征的熵的下降称为信息增益。信息增益可以用作决定一特征在解释对所述治疗的所述反应中的所述有效性的一参数。根据本发明的一些实施例,对称不决定性是可以由一特征选择演算法而使用的一算法。对称不决定性通过将多个特征归一化为[0,1]范围来补偿信息增益对于具有更多数值的多个特征的多个偏见。
子集选择算法依赖于一评估演算法及一搜索演算法的一组合,与多个特征评估演算法相似,多个子集选择算法对多个特征的多个子集进行排名。然而,与多个特征评估演算法不同,适用于本多个实施例的一子集选择演算法旨在选择对各部分的DNA数据是源自于一胎儿的所述概率具有最大影响的多个特征的所述子集,同时考虑在所述子集中包含的所述多个特征之间的冗余度。特征子集选择的好处包括促进数据可视化及理解、减少测量及存储多个要求、减少训练及利用时间以及消除分散的多个特征,以改善分类。
多个子集选择演算法的两种基本方法是将多个特征添加到一工作子集(正向选择)及从多个特征的所述当前的子集中删除(向后消除)的所述过程。在机器学习中,前向选择与具有相同名称的所述统计过程不同。通过使用交叉验证评估由一个新特征来增强的所述当前的子集的所述性能,可以找到要添加到机器学习中当前子集的功能。在正向选择中,通过将每个剩余特征依次添加到当前子集中,同时使用交叉验证评估每个新子集的所述预期的性能来构建多个子集。导致最佳性能的所述特征因为添加到当前子集后将保留并且继续进行。当所述多个其余的可用特征均未改善所述当前子集的所述预测能力时结束所述搜索,所述过程找到了一局部最优化的特征集。
向后消除以一类似的方式实现,使用后向消除功能时,当进一步减少所述特征集不会提高所述子集的所述预测能力,则结束所述搜索。本多个实施例设想了向前、向后或在两个方向上搜索的搜索算法。适用于本实施例的搜索算法的代表性示例包括但不限于穷举搜索、贪婪爬山、多个子集的多个随机扰动、多个包装算法、概率种族搜索、图式搜索、排命种族搜索及贝叶斯分类器。
一决策树是一种决策支持算法,所述决策支持算法形成了涉及考虑所述输入以做出一决策的多个步骤的一逻辑路径。
术语“决策树”是指任何类型的基于树的学习算法,包括但不限于模型树、分类树及回归树。
一决策树可用于对所述多个数据集或其关系进行分层分类,所述决策树具有包括多个分支节点及多个叶节点的树结构,每个分支节点都指定一个属性(拆分属性)及所述拆分属性的所述数值待执行的一测试(拆分测试),并且为所述拆分测试的所有可能的结果分支到其他节点。作为所述决策树的所述根的所述分支节点称为所述根节点。每个叶节点可以表示一分类(例如:一特定参数是否影响从一胎儿获取DNA数据的各个部分的所述概率)或一数值(例如:DNA数据的各个部分是源自于一胎儿的所述概率)。所述叶节点也可以包含有关所述代表的分类的附加信息,例如:测量所述代表的分类中的一可信度的一可信度得分(即所述预测的所述准确度)。
根据本发明的一些实施例可以使用的多个回归技术包括但不限于线性回归、多元回归、逻辑回归、概率回归、序数逻辑回归序数概率回归、泊松回归、负二项式回归、多项式逻辑回归(MLR)及缩短的回归。
一对数的回归或对数回归是一种回归分析,用于预测一分类因变量(一因变量可以取得一有限数量的数值,所述数值的大小没有意义,但所述数值的大小顺序可能有或可能没有意义)的所述结果。基于一个或多个预测变量,逻辑回归还可以预测每个数据点的发生概率。多个逻辑回归还包括一多项式变异。所述多项式逻辑回归模型是一回归模型,通过允许两个以上的离散结果来概括逻辑回归。也就是说,所述多项式逻辑回归模型是一个模型,用于在给定一组独立变量(可以是实值、二进制值、分类值等)的情况下预测一归类分布的因变量的多个不同可能结果的所述多个概率。对于多个二进制值变量,通常使用所述尤登(Yuden)指数决定所述0到1的多个关联之间的一界限。
一贝叶斯网络是代表多个变量及多个变量之间的条件相互依赖性的一模型。在一贝叶斯网络中,多个变量表示为多个节点,并且多个节点可以通过一个或多个链接相互连接,一链接指示两个节点之间的一关系。多个节点通常具有多个相应的条件概率表,所述多个条件概率表用于根据给定该节点所连接的其他多个节点的所述状态来决定一节点的一状态的所述概率。在一些实施例中,采用一贝叶斯最优分类器演算法将一最大后验假设应用于一新记录,以便预测所述新记录的分类的所述概率,以及根据从一训练集中获得的所述其他多个假设中的每一个来计算所述多个概率,并将所述多个概率用作多个加权因子,以便将来对DNA数据的各个部分是源自于一胎儿的一概率进行多个未来的预测。适用于所述最佳贝叶斯网络的一搜索的一演算法包括但不限于基于全局分数度量的演算法。在构建所述网络的一另一种方法中,可以使用马尔可夫毯,所述马尔可夫毯使一结点不受所述马可夫毯的边界外的任何结点的影响,所述马尔可夫毯由所述结点的父级、其子级及其子级的父级组成。
基于示例的多个技术会为每个示例生成一新模型,而不是基于由一训练集生成的多个树或多个网络(一次)进行的多个预测。
在机器学习的上下文中,术语“示例”是指一数据集中的一示例。
基于示例的多个技术通常将整个数据集存储在记忆体中,并根据一组与多个被测试的记录相似的记录来构建一模型。例如:可以通过最近邻的方法或局部加权的方法,例如:使用多个欧几里得距离来评估所述相似性。一旦选择了一组记录,就可以使用几种不同的技术(例如:所述单纯的贝叶斯)来构建所述最终模型。
多个神经网络是基于多个互连的“神经元”概念的一类演算法,在一典型的神经网络中,多个神经元包含多个数据值,所述多个数据值的每一个都会根据具有多个预定强度的多个连接以及与每个特定神经元的多个连接的所述总和是否满足一预定的阈值来影响一连接的神经元的所述数值。通过决定多个适当的连接强度及多个阈值(也称为训练的一过程),一神经网络可以实现多个图像及多个字符的有效的识别。通常地,将所述多个神经元分为几层,以使多个群组之间的多个连接更加明显,并且得到多个数值的每个计算。所述网络的每一层可能具有不同数量的神经元,并且所述多个神经元与所述输入数据的多个特定的质量可能有关或可能无关。
在被称为一完全连接的神经网络的一实现方式中,在一特定层中的所述多个神经元中的每一个皆连接并提供输入值到所述下一层中的所述多个神经元,然后将所述多个输入值加总,并且将所述加总及与一偏差或阈值进行比较。如果所述数值超过一特定神经元的所述阈值,则神经元将保留一正值,所述正值可用作所述下一层的神经元中的多个神经元的输入。所述计算将在整个神经网络的各个层继续进行,直到所述计算到达一最后一层。此时,可以从所述最后一层中的所述多个数值读取所述神经网络例程的所述输出。与多个完全连接的神经网络不同,卷积神经网络通过将多个数值的一阵列与每个神经元而不是单一值相关联来进行操作,所述后续的层的一神经元数值的所述转换是从乘法到卷积。
根据本发明的一些的实施例使用的所述机器学习程序是一被训练的机器学习程序,可选地及优选地是一深度学习程序(例如:一卷积神经网络),所述深度学习程序提供与馈送到所述深度学习程序的所述多个参数非线性相关的输出。
预计在从本申请至一专利寿命到期的期间,将开发许多相关的机器学习程序,并且机器学习程序一词的范围意在包括所有类似的先前技术。
根据本发明的一些实施例,可以通过馈送如上面进一步详细描述的育有一基因型胎儿的一群对象中的每个的DNA数据以及可选地及优选地元数据来训练一机器学习训练程序可选地且优选地是深度学习程序,例如但不限于:一卷积神经网络。当使用一卷积神经网络时,所述网络的所述输入通常地(但不一定)是多个多维张量的形式。例如:每个张量可以包括对应于所述基因组中一特定基因座的输入数据。在多个优选的实施例中,所述张量是覆盖一候选单核苷酸多态性(SNP)(例如:以所述SNP为中心)的多个读数的一堆叠(pileup),所述张量的前两个维度可以例如对应于所述堆叠的长度及读段的数量,并且所述张量的一个或多个其他维度可以对应于所述SNP的元数据。在所述训练阶段,每个张量都与一标签相关联,所述标签包含使用一侵入性测试发现的所述真实的胎儿基因型。
一旦所述数据被馈送,所述机器学习训练程序就会生成一被训练的机器学习程序,然后可以使用所述机器学习程序而无需重新训练,也不需要使用向所述机器学习程序馈送一侵入性测试发现的多个胎儿基因型。所述被训练的机器学习程序可以提供一部分所述母源性DNA数据是来自一胎儿的概率,及/或可以将所述父母中的至少一个具有一突变的一位点分类为胎儿或母体。
优选地,将所述被训练的机器学习程序用作一端对端程序,在这种情况下,将所述DNA数据馈送到所述被训练的机器学习程序,并且指示一位点是从一胎儿中获得的所述概率的一输出,或者更优选地可以从被训练的机器学习程序中接收所述父母中至少一个具有一突变的多个位点被分类为胎儿或母亲的一输出。
替代地,被训练的机器学习程序可以用于其他技术的补充,例如用于重新校准。例如:在其中在16中通过一贝叶斯程序进行所述分类的多个实施例中,所述被训练的机器学习程序可以用于重新校准所述贝叶斯程序的所述输出。
进一步设想了在多个实施例中,被训练的机器学习程序的所述输出用于再训练所述机器学习程序,从而允许随着时间的过去提高所述准确度。
根据本发明的一些实施例,可以通过一计算机或一服务器-客户端计算机配置来执行胎儿基因分型,如现在将参考图16所说明的。
图16示出了具有一硬件处理器32的一客户端计算机30,所述硬件处理器通常包括一输入/输出(I/O)电路34、一硬件中央处理单元(CPU)36(例如:硬件微处理器)及一硬件存储器38。所述硬件存储器38通常包括挥发性记忆体及非挥发性记忆体。CPU 36与I/O电路34及存储器38通信。客户端计算机30优选地包括与处理器32通信的一用户界面,例如:一图形用户界面(GUI)42。I/O电路34优选地以适当的结构的形式与GUI 42进行信息通信。还示出了一服务器计算机50,所述服务器计算机50可以类似地包括一硬件处理器52、一I/O电路54、一硬件CPU 56、一硬件存储器58。客户端30及服务器50的计算机的多个I/O电路34及54优选地作为多个收发器运行,所述多个收发器通过一有线或无线通信相互通信信息。例如:客户端30及服务器50的计算机可以经由例如一局域网(LAN)、一广域网(WAN)或因特网之类的一网络40进行通信。在一些实施例中,服务器计算机50可以是通过所述网络40与客户端计算机30通信的一云计算设施的一云计算资源的一部分。
GUI 42及处理器32可以集成在同一壳体内,或者所述GUI 42及处理器32可以是彼此通信的多个独立单元。GUI 42可以可选地及优选地是包括一专用CPU及多个I/O电路(未示出)的一系统的一部分,以允许GUI 42与处理器32通信。处理器32向GUI 42发布由CPU 36生成的图形及文本输出。处理器32还响应于用户输入从GUI 42接收与由GUI 42生成的多个控制命令有关的多个信号。GUI 42可以是本领域中已知的任何类型,例如但不限于:一键盘及一显示器、触摸屏等。在多个优选实施例中,GUI 42是例如一智能电话、一平板电脑、一智能手表等的一移动设备的一GUI。当GUI 42是一移动设备的一GUI时,所述移动设备的所述CPU电路可以用作处理器32,并且可以可选地及优选地通过执行多个代码指令来执行所述方法。
客户端30及服务器50的计算机可以进一步分别包括一个或多个计算机可读存储介质44、64。多个介质44及64优选地是多个非暂时性存储介质,所述介质44及64存储用于执行本多个实施例的所述方法的多个计算机代码指令,并且多个处理器32及52执行所述多个代码指令。可以通过将所述相应的代码指令加载到所述多个相应的处理器32及52的所述相应的执行存储器38及58中来运行所述多个代码指令。多个存储介质64优选地还存储一个或多个数据库,所述数据库包括如上面进一步详细描述的带有多个心理学注释的嗅觉签名的数据库。
在操作中,客户计算机30的处理器32接收DNA数据,并且可选地及优选地如上所述接收元数据,可以例如从存储介质44读取所述数据。处理器32通常通过网络40将所述数据发送到服务器计算机50。介质64可以存储如上所述的用于对胎儿进行基因分型的一程序(例如:一深度学习程序及/或一贝叶斯程序)。服务器计算机50可以访问多个介质64,向所述存储的程序馈送从客户端计算机30接收的所述数据,并且从所述程序接收指示所述胎儿的所述多个基因型或指示所述数据的一部分(例如:一突变位点)是源自于胎儿的所述概率的一输出。服务器计算机50还可以将所述获取的输出发送到客户端计算机30,并且客户端计算机30可以在GUI 42上显示所述信息。
替代地,介质44还可以存储如上面进一步详细描述的对所述胎儿进行基因分型的所述程序,在所述情况下,整个方法可以由计算机30执行。具体地,计算机30可以将从介质44接收的所述DNA数据存储到存储器38中,并访问介质44以获得对所述胎儿进行基因分型的所述程序,将被存储在存储器38中的所述数据馈送到所述程序,并从所述程序接收指示所述胎儿的所述多个基因型或指示所述数据的一部分(例如:一突变位点)是源自于胎儿的所述概率的一输出。然后,计算机30可以在GUI 42上显示所述信息。
如本说明书所用的术语“约”是指±10%。
本说明书中所用的词语“示例性(exemplary)”的意思是“用作示例、例子或例证”。以“示例性”描述的任何实施方式不必被理解为比其他实施方式优选或有利和/或排除来自其他实施方式的特征的并入。
本说明书中所用的词语“可选择地(optionally)”的意思是“在一些实施例中提供,而在其它实施例中不提供”。任何本发明的特定实施例可以包括多个“可选择的”特征,除非此类特征相冲突。
术语“包括(comprises)”、“包括(comprising)”、“包括(includes)”、“包含(including)”、“具有(having)”及其词形变化是指“包括但不限于”。
所述术语“由…组成(consisting of)”意思是“包括及限于”。
所述术语“主要由…组成(consisting essentially of)”意思是所述组合物、方法或结构可以包括额外的成分、步骤和/或部件,但只有当额外的成分、步骤及/或部件实质上不改变所要求保护的组合物、方法或结构的基本特征及新特征。
本文所用的单数形式“一(a)”、“一(an)”以及“所述(the)”除非在上下文另有明确指出,否则本发明可包括复数个参考物。例如:术语“一化合物(acompound)”或“至少一化合物(at least one compound)”可以包括多个化合物,包括它们的混合物。
在整个本申请中,本发明的各种实施例可以以一个范围的型式存在。应当理解,以一范围型式的描述仅仅是因为方便及简洁,不应理解为对本发明范围的硬性限制。因此,应当认为所述的范围描述已经具体公开所有可能的子范围以及所述范围内的单一数值。例如,应当认为从1到6的范围描述已经具体公开子范围,例如从1到3,从1到4,从1到5,从2到4,从2到6,从3到6等,以及所数范围内的单一数字,例如1、2、3、4、5及6,此不管范围为何皆适用。
每当在本文中指出数值范围,是指包括所指范围内的任何引用的数字(分数或整数)。多个短语:第一指示数字及第二指示数字“之间的范围”及第一指示数字“至”第二指示数字“的范围”在本文中可互换,并指包括第一及第二指示数字,及其之间的所有分数及整数。
本文中所使用的术语“方法”是指用于完成一特定任务的方式(manner)、手段(means)、技术(technique)及程序(procedures),所述给定的任务包括但不限于已知的或是从已知的方式、手段、技术或步骤很容易地由化学、药理、生物、生化及医学领域的从业者开发所述多个方式、手段、技术以及程序。
如本文所用,术语“治疗”包括终止、基本上抑制、减慢或逆转病症的进程,基本上改善病症的临床或心理症状或基本上预防病症的临床或心理症状的出现。
当参考多个特定的序列表时,所述参考应当被理解为还包括基本上对应于所述多个特定的序列表的互补序列的多个序列,包括:由于例如定序错误、克隆错误或导致碱基置换、碱基缺失或碱基增加的其他改变而导致的多个较小的序列变异,前提条件是所述多个变异的频率为小于五十分之一个核苷酸、小于一百分之一个核苷酸,替代地,小于二百分之一个核苷酸、小于五百分之一个核苷酸,替代地,在小于一千分之一个核苷酸,替代地,小于五千分之一个核苷酸,替代地,小于一万分之一个核苷酸中。
应当理解,本发明的某些特征,为了清楚阐明,描述在多个独立的实施例的上下文中,也可以是在一单一实施例中以组合提供。相反,本发明的各种特征,为了简明,在一单一实施例的上下文中描述,也可以单独或以任何合适的子组合或以适合于本发明的任何其它描述的实施方式来提供。在各种实施例的上下文中描述的部分特征不应被认为是那些实施例的主要特征,除非所述实施例在没有这些组件的情况下不运作。
如上文描绘和下文权利要求部分所要求的本发明的各种实施例和各个方面可在下文的多个实施例中找到实验支持。
多个示例:
现在参考以下多个示例,连同以上的多个描述一起以一非限制性方式说明本发明的一些实施例。
非侵入性父母诊断(NIPD)是一项针对所述胎儿的多个遗传性变异的一检测,所述非侵入性父母诊断(NIPD)基于血浆中自由循环的多个细胞游离DNA(cfDNA)片段。在怀孕期间,所述母体的血浆中的细胞游离DNA包含起源于多个胎盘细胞的细胞游离胎儿DNA(cffDNA)。细胞游离胎儿DNA的所述含量(也称为胎儿分数)在怀孕的前三个月末约占所述细胞游离DNA的10%,在随后的三个月中增加。可用的NIPD目前主要用于多个染色体异常(例如:唐氏综合症)。
点突变的NIPD是一项更具挑战性的任务,因为与一整个染色体相比,覆盖一给定基因组位置的细胞游离胎儿DNA的所述含量要相当地低。在仅有父亲为一携带者的多个突变中,可以将所述母体的血浆中的所述外来(非母体)的等位基因的存在建模为二项分布;如果所述母体的血浆中足够多个DNA片段包含所述外来的等位基因,则可以推断出所述外来的等位基因是遗传的。因此,在所述位置中对胎儿的一突变的检测被认为更为直接明瞭。在只有母亲是杂合子的多个突变中,无论所述遗传的等位基因的所述识别如何,两个等位基因都大量存在于所述母亲的血浆中,解决所述问题的常用方法是测量每个等位基因的数量。如果所述多个等位基因具有相等的数量,则认为所述胎儿是所述突变的一携带者,即一杂合子。如果存在一等位基因失衡,则所述胎儿与一过量存在的所述等位基因是纯合的。
测量多个细微的等位基因失衡需要一超精确的定量方法,例如:一数字聚合酶链反应(PCR)。所述多个发明人意识到所述方法需要针对每种情况进行一定制设计,因此,所述方法不适合同时测试大量突变。另一种选择是使用次世代测序(NGS),所述次世代测序是一种不太准确的方法,但是以一非常深的覆盖面来进行所述次世代测序(即每个位置均被读取了多次),并且使用了可以减少多个伪像的多个特殊制备方案,几乎没有方法被建议用于多个点突变的基于NGS的全基因组NIPD(Chan等人2016;Fan等人2012;Kitzman等人2012)。
本多个发明人发现,对于母亲来源的多个SGD,敏感性造成所述检测被限制为一次一个遗传性疾病的一挑战。下面的示例1提供了一种用于多个单基因疾病的所述NIPD的一贝叶斯程序,所述方法独立于遗传方式及父母来源。被测序的父母及胎儿数据被用于校准及验证所述模型。示例1示出了考虑到源自于胎儿及母亲的细胞游离DNA的片段长度分布的多个差异导致增加的准确度。示例1的所述模型扩展到遗传的插入与缺失(indels)的预测。示例1还示出了使用来自于多个先前分析的家族的数据并通过一机器学习演算法重新校准所述模型的所述多个后验概率,可纠正一大量的基因分型错误。在示例1中,本多个实施例的所述方法成功地应用于预测所述父母为一突变的多个携带者的一妊娠中的先天性氯化物腹泻的遗传。本实施例证实了下一代测序(NGS)可同时用于多种单基因疾病的NIPD,所述实施例表明了本多个实施例的所述方法可以用作多个SGD的所述NIPD的一通用框架。
通常地,NIPD可通过分析所述母体的血浆中的细胞游离DNA来实现,所述血浆中含有源自于所述胎盘的细胞游离胎儿DNA,所述细胞游离胎儿DNA的主要用途是识别多个染色体异常,例如:三体症(唐氏综合症)。多个其他临床应用是胎儿性别决定及恒河猴D基因分型。可以通过各种方式实现SGD的遗传诊断,从所述表覝型描述及一连锁分析、通过例如用于多个已知的突变的聚合酶链反应(PCR)及多个DNA微阵列等多个不同的实验室检测到用于确认多个结果的桑格定序及用于一更深入的调查的NGS。两个已知的工具是基于NGS的全外显子组测序(WES)及全基因组测序(WGS)。尽管WGS的成本仍然很高,并且WGS的多个结果的多个含义尚待研究,但WES覆盖了大约所述基因组中的2至3%且成本较低,故WES仍经常被使用。然而,即使在多个外显子组变异中,WGS也比WES更可靠,并且WGS具有解决多个结构变异的能力,因此变得越来越流行。被怀疑患有多个遗传性疾病的多个婴儿的WES更有可能对医疗照护具有影响,通过羊膜穿刺术获得的DNA的WES在一些情况下可以帮助进行所述产前诊断。
本多个发明人发现,尽管NGS在多个单基因疾病的所述NIPD中的所述应用已被证明是可行的,但仍可以做出一些改进。不同于在细胞游离DNA中的父源传递的等位基因的识别被认为直接明瞭,母源传递的等位基因构成了一更大的挑战,因为在所述母亲为杂合子的多个位点中,两个等位基因都存在于她的血浆中。在所述多个情况下,常规技术无法决定所述多个等位基因中的何者是遗传的。当前的解决方案是寻找一等位基因失衡(当所述胎儿是纯合子时出现的一等位基因的一略高的数量)。然而,由于细胞游离DNA的多个数量少,甚至细胞游离胎儿DNA的多个数量更少,因此所述等位基因失衡的执行仅限于例如数字PCR等多个超精确设备。再者,所述方法是不可扩充的;。当测试了更多个基因组位点时,所述方法变得不太可行,因此,没什么帮助。也可以使用NGS,而NGS需要对每个位点进行一非常深入的覆盖。
已知一种使用WES来提供更深入的覆盖范围的一技术[Fan,H.C.等,所述胎儿基因组的无创产前测量,自然487,320至324(2012)]。在这项技术中,当分别在第二及第三妊娠期使用深度WES(221x及631x)时,一高比例的所述胎儿外显子被重建。在所述分析之前应用了严格的数据过滤。在对一胎儿进行基因分型的另一种尝试中,使用WGS对细胞游离DNA进行了一深度覆盖(270x)的测序,并且在所述母亲为杂合子且没有对所述父母进行单倍型的基因座中对每个位点进行了一顺序概率比测试[Chan及K.C.A.等人,第二代非侵入式胎儿基因组分析揭示了新生突变、单碱基亲本遗传及多个优选的DNA末端,美国国家科学院学报,201615800(2016),数字对象识别符(doi):10.1073/pnas.1615800113]。尽管所述研究显示出良好的准确度,但本发明人发现了所述技术的一些局限性。首先,所述被测序的样品来自第三妊娠期,其中细胞游离DNA的所述数量及在所述细胞游离DNA中的细胞游离胎儿DNA的数量都很高。第二,所述应用的方法没有使用有关所述父源性遗传的可用信息。第三,尚不清楚在对一单一位置进行基因分型时一顺序测试是否具有一优点,因为所述情况下的所述数据不是累积性。另外,在所述研究中,准确度是从一相对较低的仅6.5x 10
所述多个发明人发现可以帮助改善非侵入性胎儿基因分型的一方法,可以依赖于多个胎儿及母亲特征的多个固有差异,所述差异中的一个是所述实际的细胞游离DNA片段及细胞游离胎儿DNA片段。例如:源自于胎儿的多个片段通常较短,并且所述源自于胎儿的多个片段的大小分布的所述模式指示与核小体的定位的一关系,进行多次尝试通过施加一硬阈值设置以利用多个大小差异来处理多个染色体异常,以丰富细胞游离胎儿DNA。然而,所述多个发明人发现由于所述两个大小分布大量重叠,因此,所述阈值可能导致相关信息的丢失。
下面的示例1示出了通过使用深度NGS及一改进的演算法,可以以非侵入性的方式准确地检测所述胎儿中的多个小变异。示例1使用一贝叶斯模型证明了在即使所述母亲或父母双方都是一突变的多个携带者的多个位置以及在多个小的插入与缺失(indels)中也能准确检测出所述胎儿中的多个小变异,所有所述多个小变异都被认为更难以进行基因分型。根据多个简单的孟德尔遗传定律,使用所述亲本基因型计算所述先验概率,并且所述似然函数是基于每种可能的胎儿基因型中的每个细胞游离DNA片段的多个支持。所述多个片段长度也被用于所述似然函数,因为先前描述的多个胎儿片段及母源性片段的长度分布不同,所述多个长度分布及所述胎儿分数(即所述细胞游离DNA内的细胞游离胎儿DNA的所述比率)皆可以根据一给定的细胞游离DNA样品进行经验计算,所述贝叶斯模型也由一机器学习重新校准程序进行辅助。
下面的示例2提出了一种基于一深度学习程序的端到端方法,所述深度学习程序可以代替示例1中使用的所述贝叶斯模型及所述机器学习重新校准程序。示例2中介绍的所述端到端方法的所述多个优点中的一个是所述端到端方法能够自动对多个系统错误进行建模,所述能力在难以解读的数据(例如:低深度测序)中很有用。随着所述两种编程方法的易于使用以及硬件价格的下降,深度学习正在迅速变得可用。
示例1:
所述实施例描述了用于一种非侵入性产前变异侦测的技术,本发明人将所述技术称为霍巴里(Hoobari)。霍巴里技术基于一贝叶斯演算法,所述贝叶斯演算法单独使用每个读段的所述信息,可以以一模块化的方式来微调所述技术,而不必重写整个模型,这样的一示例是基于一机器学习的多个概率重新校准步骤,所述技术使用片段大小差异以改善胎儿基因分型,这对于所述母亲是杂合子的多个具有挑战性的基因座特别有用。可以对本示例中示出的所述技术进行广义化,以允许预测多个小的插入与缺失(indels)的所述遗传。本发明人使用两个第一妊娠期的个案的深度WES(>600x)及另一个第一妊娠期的个案的深度WGS(310x)证明了所述技术解决基于NGS的所述诊断的能力,从而通向一广泛的SGD的NIPD。
多个材料及方法:
考虑到所述母亲为杂合的多个单核苷酸多态性(SNP),由于很难决定在所述多个位点中的一片段是胎儿还是母源性,因此,根据所述片段的大小计算每个片段是胎儿的概率,并将所述多个概率用于一贝叶斯分类模型中。
使用所述多个父母在多个不同的等位基因是纯合的多个位点,创建了两个经验大小分布:一胎儿经验大小分布及一母亲经验大小分布。在所述多个位点中,显示所述父源性等位基因的一细胞游离DNA片段很可能是胎儿的。计算所述总胎儿分数,所述总胎儿分数为所有母源性细胞游离DNA中的细胞游离胎儿DNA的所述分数。然后,使用具有所述相同长度的所有片段来计算每个片段大小的一胎儿分数。当将所述贝叶斯模型应用于一特定的基因组的感兴趣的位点时,所述按大小划分的胎儿分数被用于计算覆盖所述位点的所有的所述多个片段中的每个片段是胎儿的概率。以所述方式,较短的片段通常具有较高的胎儿概率,并且可以避免严格的尺寸阈值。
如图1示意性地说明了霍巴里(Hoobari)用于非侵入式产前变异检查的流程,所述代码送回三个后验概率,每个可能的胎儿基因型各一个后验概率:与所述参考的等位基因纯合(0/0)、杂合(0/1)、及与所述变异的等位基因纯合(1/1)。每个位点的所述预测基因型是概率最高的所述基因型,一纯胎儿样品(例如:羊水、绒毛膜或脐带血)所发现的多个胎儿变异被用作多个真正的变异。
所述流水线与常规的变异侦测工作流的不同之处在于使用从所述父母的一初始基因分型获得的现有父母测序数据,以高可信度计算了所述多个先验概率。另外,细胞游离DNA是两个相似基因组的一不平衡混合物。因此,在用于计算所述贝叶斯模型中的所述多个似然性的所述专用技术中,所述流水线也不同于常规的变异侦测工作流,所述技术使用所述细胞游离DNA片段长度分布,所述细胞游离DNA片段长度分布是在一预处理步骤中计算得出的。
以下是对所述研究的一更详细描述。
处理多个以前的研究中的多个家庭:
本实施例的所述技术是在多个先前研究中测序到不同覆盖深度的四个家族家系(trios)的全基因组数据上进行测试,并且所述多个细胞游离DNA内的所述胎儿分数具有变化。所述多个家族被称为G1、G2、G3及G4。如上面的Chan及K.C.A等人及Kitzman及J.O.等人,人类胎儿的非侵入式全基因组测序,科学转化医学,4,137ra76(2012)对来自于G1至G2家族及G3至G4家族的多个样品进行收集及测序。
样品收集及DNA提取:
在获得知情同意书的情况下,在第11周期间收集了来自每个家庭的多个样品,使用所述MagNA纯紧凑型核酸分离试剂盒I-大体积(罗氏生命科学)的所述DNA组织操作流程从绒毛膜绒毛取样(CVS)中提取DNA。使用多个2-4乙二胺四乙酸(EDTA)管收集外周母体血液,通过在4℃以1600×g离心10分钟将血浆与血液分离,然后将所述血浆在室温下再次以16,000x g离心10分钟,以除去任何残留的细胞。使用所述QIAamp循环核酸试剂盒(Qiagen)进行细胞游离DNA的提取。使用阿根考特(Agencourt)AMPure XP磁珠(贝克曼库尔特公司)以细胞游离DNA体积的一2倍比例去除细胞游离DNA纯化产所生的多余盐类,根据生产商的多个指示使用包括(i)血沉棕黄层分离及(ii)使用简塔(Gentra)纯净基因血液试剂盒(凯杰(Qiagen))的一操作流程进行从所述母体的血沉棕黄层中的多个白细胞中提取纯净的母源性DNA,同样地收集及纯化纯净的父源性DNA。
文库构建及测序:
根据制造商的多个指示使用TruSeq DNA无PCR的文库构建试剂盒(因美纳(Illumina))对经过WGS的多个样品进行文库构建,随后使用所述HiSeq X十系统(Illumina)以151个碱基对(bp)的多个双端(paired-end)读段进行测序。
对于多个经过WES的样品,根据制造商的多个指示使用绝对选择(SureSelect)V5外显子组试剂盒(安捷伦(Agilent))进行文库构建,通过将被构建的基因组DNA与互补RNA探针杂交来实现富集,然后使用HiSeq 4000(因美纳)以101个碱基对的双端读段进行测序。
多个细胞游离DNA样品在文库构建期间中没有片段化,并且在两个步骤进行测序:(1)使用HiSeq 4000(因美纳)以101个碱基对的双端读段达到一要求的50x覆盖率;(2)使用NovaSeq(因美纳)以151个碱基对的双端读段达到一要求的950x覆盖率。
与所述基因组比对:
使用具有多个默认参数的伯罗斯惠勒(Burrows-Wheeler)第0.7.834个版本将多个读段与基因组参考联盟人类构建37(GRCh37/hg19)比对,从PCR克隆性或多个光学重复得到的多个重复读段,并且将配对到多个位置的多个读段排除在下游分析之外。
纯基因组测序数据的变异侦测:
使用免费贝叶斯(Freebayes)软件第1.1.0-3-g961e5f335个版本应用多个默认参数,可以识别出多个单核苷酸替代及多个小的插入与缺失。首先,免费贝叶斯将父母的所述比对测序数据一起运行,然后使用在所述多个亲本基因组中识别的所述多个变异位点在所述CVS样品的所述比对数据上运行。由于多个被报告的变异未被过滤,因此所有被报告的多个SNP及多个插入与缺失均被保留用于下游分析。
细胞游离DNA数据的预处理:
免费贝叶斯仅在多个亲本基因组中识别的多个变异位点上对所述细胞游离DNA样品运行,使用霍巴里(Hoobari)将每个读段观察到的所述等位基因以及所述读段的插入片段大小都保存在一单独的数据库中。
非侵入性胎儿变异侦测:
使用亲本变异及细胞游离DNA预处理结果数据库作为输入来运行霍巴里,所述输出是一标准的变异侦测格式(variant call format,VCF)文件,使用几种专用于VCF操作的软件(例如:vcflib及vcftools)对所述多个结果进行所述分析。
贝叶斯非侵入式的基因分型:
在每个感兴趣的位点,应用一贝叶斯计算。对于每个可能的胎儿基因型:
其中G是胎儿基因型,而G
一读段r
确认E1家族中的所述突变:
在一患病后代的所述前一胎出生后,使用桑格测序在父母双方中发现了SLC26A3基因的一有害的无意义突变(NM_000111.2,c.559G>T),所述突变会导致先天性氯化物腹泻(CCD),所述先天性氯化物腹泻以常染色体隐性遗传。使用所述CVS样品的Sanger测序证实了所述研究中测序的所述胎儿与所述突变纯合。
多个结果:
利用多个片段大小进行胎儿基因分型
在每个家族中,对所有个体的所述基因组DNA进行测序以覆盖范围在25x到60x之间,所述多个细胞游离DNA样品被测序到多个深度为270x、195x、78x及56x(请参见下面的表1)。计算出的所述总胎儿分数分别为32.4%、24.9%、14%及8.7%。因此,根据两种覆盖程度将所述四种情况分类:高(G1至G2)及低(G3至G4)。
表1:多个先前研究的多个样本的汇总
针对每个家族执行本实施例的所述方法,以使用所述多个后验概率与所述准确度之间的所述关系来测试所述方法演算法的所述校准,并且决定添加多个片段大小是否增加所述准确度。
为了测试有无片段大小的所述准确性,所述方法执行一次,将所述总胎儿分数作为每个片段的一固定参数,然后再次使用取决于所述片段大小的一胎儿分数。在所有四个家族中,所述方法均显示出令人满意的校准效果,并且片段大小信息的添加可提高准确度。图2A-C显示了在多个SNP中只有所述母亲是杂合子(图2A)、只有父亲是杂合子(图2B)或父母都是杂合子(图2C)的多个SNP中,多个家族G1至G4的所述准确度与所述后验概率之间的所述关系。示出了所述准确度是最大的后验概率的所述阈值的一函数,所述函数表示所述多个预测的决定性水平。一校准后的模型应显示出一平滑的上升曲线。对于多个低覆盖率情况(多个家庭G3至G4,图2A至2C,参见图2D中的多个颜色代码),观察到较低的校准,但是所述片段大小的所述使用显示出校准的一改进。
非侵入性产前的插入与缺失侦测:
本多个实施例的所述方法也被用于父母插入与缺失侦测。再次,使用所述总胎儿分数作为一固定参数,然后以每一片段大小的胎儿分数执行所述方法。图3A至3C(图3D中的多个颜色代码)示出了在所述母亲是杂合子的多个插入与缺失位点处的所述模型的校准,所述精确度作为每个位点的所述最大后验概率阈值的一函数被示出在多个插入位点中,对于G1至G4的情况,所述多个插入位点是仅有母源性的杂合子(父源性纯合子)(图3A)或仅有父源性杂合子(母源性纯合子)(图3B)或两个亲本都是杂合子(图3C)。具有一每个片段大小的胎儿分数的多个方法执行以红色显示,具有一固定胎儿分数的多个方法执行以蓝色显示。
在一固定的胎儿分数的情况下,所述多个低覆盖率的多个家庭显示出较低的准确度,并且所述校准结果不一致,而所述高覆盖率的家庭显示出更高的准确度及非常好的校正结果。然而,当使用所述片段大小信息时,在准确度及校准上都实现了一相当大的提高(图3A至3C)。当所述方法应用于所述父亲与一插入与缺失是杂合的多个位点,而所述母亲与所述参考或所述插入与缺失是纯合的多个位点时,与所述母源性杂合的多个插入与缺失的位点相比,所述准确度更高(图3A至3C)。以所述大小信息,所有家族都表现出一更好的校准效果,而在所述高覆盖率的家族中的所述准确度被改进。
模拟数据分析:
使用模拟数据检查了本多个实施例的所述模型在具有高测序深度的多个低胎儿分数下的多个稳健性,从G1家族中对具有胎儿分数的多个变化值的多个细胞游离DNA样本的六个三重复进行了二次采样,同时保持其测序深度。根据多个测序深度进一步对每个胎儿部分进行分类,在具有所述最大深度的所述最高胎儿分数下,结果显示出每一基因座类别的准确度很高:仅母源性杂合子的基因座为96.0%、仅母源性杂合子的基因座为99.1%、父母均为杂合子的基因座为91.1%(图4A至4C)。对于在所述第一妊娠期中更常见的多个胎儿分数值(即10至15%),多个相同类别的最大测序深度的所述准确度分别为88.4至92.4%、99.1%及82.8至87.1%。
使用深WES/WGS分析的3个第一妊娠期的案例
对三个家族进行了测序(参见下面的表2),并将所述三个家族的数据输入到本实施例的所述方法中。在两种情况下,为了获得对所述测序区域的一较深的覆盖度,使用了WES(约占所述基因组的2至3%)。在第三个情况下,使用了深度WGS。在E1家族中,所述父母是导致先天性氯化物腹泻(CCD)(常染色体隐性疾病)的一突变的多个携带者。
表2:
多个测序样品的汇总
*中位数,按目标。
与之前相似,在所述母亲是杂合的多个位点的多个SNP及插入与缺失上测试了本多个实施例的所述方法,图5A至5F(参见图5G中的多个颜色代码)示出了多个家族E1、E2及G5的多个SNP位点(图5A至5C)及多个插入与缺失位点(图5D至5F)的校准,示出了作为所述最大的后验概率的阈值的一函数的所述准确度。
E1、E2及G5家族的所述细胞游离DNA样本中计算出的所述多个胎儿分数分别为15.8%、12.8%及18.5%。图5A至5F证明了所述方法已被很好地校准。此外,即使所述总准确度受到限制,通过在所有情况下增加片段长度信息也可以实现一改进。在E2家族中,所述校准也得到了一改进。所述演算法大体上针对多个SNP进行了更好的校准,尽管在使用WGS来验证多个变异的E2家族中的所述多个结果较低。对于多个插入与缺失,所述模型在所述E2家族中具有一很好的校准,G5家族在所有SNP及插入与缺失的多个分类中均表现出多个最高的结果。
在所述多个预测的SNP中,成功预测了SLC26A3中的所述突变的所述胎儿基因型,所述预测示出了所述胎儿与所述突变的等位基因是纯合的,这一结果与绒毛膜绒毛取样(CVS)的所述WES相匹配,进一步使用桑格测序验证了这一点。当包含了与多个片段长度有关的信息时,所述预测的所述确立性更高。在所述相关的位点,具有长度信息的所述后发概率为61%,而没有所述长度信息的所述后发概率为56%。正如预期,在E2家族中未检测到所述突变。
基于机器学习的变异概率重新校准
在所述步骤中,使用一新训练的机器学习模型来改进每一新处理的样本的所述多个结果,所述模型中的所述多个特征取自于当所述父母及所述细胞游离DNA进行基因分型时可用的所述元数据(请参见附件1中的表A.1)。所述测序深度及胎儿分数最高的G1家族被选为所述第一训练集,G2家族被随机划分,并且所述多个变异的75%被用作针对经过G1训练的多个不同的模型的一验证集。发现了所述随机森林演算法具有多个最好的结果。已经发现在仅有产妇及双杂合的多个SNP中,应该使用在同一类别内的基因座进行所述训练,例如:旨在改善仅有多个母源性杂合的SNP的所述多个结果的一模型将仅会在仅有母源性杂合的多个SNP上进行训练。在仅有父源性杂合的多个SNP中,应使用所有基因座中的父母中的至少一个是杂合的进行训练。应当对同一类别中的多个SNP及多个插入与缺失来训练多个插入与缺失。
所述选定的模型在G2的所述剩余25%上进行了一测试,再次对合并的多个家族G1及G2训练了所述相同的模型,并且在G5家族上进行了一测试,即在一不同的数据集上进行了测试。在所有类别的基因座中,所述接收器工作特性曲线(AUC)下的所述面积都得到了改善,并且准确度几乎在所有情况下都得到了改善(图6A至6F)。在父母均为杂合子的基因座并且原本表现出较低的准确度的一情况中观察到显著的改善。还发现了在重新校准之后,所述多个预测概率更好地表示所述实际的准确度,即当所述后验概率为0.9时,所述预期的准确度也为~0.9。此外,在每个预测概率阈值处的所述准确度都得到了显著改进,表明基因座的过滤确实可以使用重新校准来执行,而不是为多个特定的特征设置多个严格的阈值。例如:在0.7的所述阈值处,从722,630个仅有母源性杂合基因座的一总数、1,358,503个仅有父源性杂合基因座及358,114个双杂合基因座中,G5家族的所述准确度提高到比多个SNP高98.3-99.8%。从42,726个仅有母源性杂合基因座、142,577个仅有父源性杂合基因座及20,388个双杂合基因座,将插入与缺失预测准确度提高到94.3至97.1%。还发现衍生自霍巴里(Hoobari)的多个特征,例如:所述先验概率及所述后验概率,所述可能性及所述预测的基因型具有最高的重要性,其次是与所述细胞游离DNA中的所述等位基因平衡相关的多个特征。
图7A至7F示出了所述模型的准确度的校准(图7G中的多个颜色代码)。
示出的是变异重新校准前后的所述准确度作为所述预测概率的一函数。所述“完全校准”线表示一种所述概率被分配到一预测完美地描述了正确的多个概率(就准确度而言)的情况,每个图的所述下方的窗格中示出的是每个分箱中的基因座的边缘分布。图7A对应于多个仅有母源性杂合的SNP,图7B对应于多个仅有父源性杂合的SNP,图7C对应于多个父母皆杂合的SNP,图7D对应于多个仅有母源性杂合的插入与缺失,图7E对应于多个仅有父源性杂合插入与缺失,图7F对应于多个父母皆杂合的插入与缺失。
图8A至8F示出了在重新校准之后的所述模型的性能(图8G中的多个颜色代码),图8A至8F示出了执行基于机器学习的变异重新校准以过滤多个变异的能力。如图2A至2D、3A至3D及5A至5G所示,在本说明书中示出了一后验概率高于所述x轴的所述多个阈值的基因座的多个准确度。在变异重新校准前后都会提供多个发现。每个类别的所述总准确度是所述x轴上的最左点的所述准确度。在每个图示的所述底部示出了用于计算每个阈值的所述准确度的所述多个总计数。如图8A对应于多个仅有母源性杂合的SNP,图8B对应于多个仅有父源性杂合的SNP,图8C对应于多个父母杂合的SNP,图8D对应于多个仅有母源性杂合的插入与缺失,图8E对应于多个仅有父源性杂合的插入与缺失,图8F对应于多个父母杂合的插入与缺失。
图9A至9G分别是多个家族G1、G2、G3、G4、E1、E2及G5的片段长度分布的多个密度图。
图10示出了使用桑格测序所获得的E1家族的SLC26A3基因中的所述突变的多个确认结果,第一行示出了与所述参考等位基因(G)是纯合的一对照组,第二行示出了所述父亲是所述变异的等位基因(T)的一携带者,第三及第四行显示了使用一正向引物(第三行)及一反向引物(第四行)的所述胎儿的所述多个结果,所述胎儿与所述变异的等位基因是纯合的。
讨论:
在本示例中,使用本多个实施例的所述方法证明了非侵入性胎儿的基因分型,本示例证实了母源性及胎儿来源的多个片段之间的所述多个大小差异改善了基于细胞游离DNA的胎儿基因分型,所述多个结果在目前构成了最大的识别挑战的所述母亲是杂合的多个位点上是突出的,本示例还证明了本多个实施例的所述方法预测多个胎儿的插入缺失的所述能力,多个插入与缺失是第二种最常见的变异类型,并且可能是有害的,特别是当它们影响所述阅读框时。
所述贝叶斯程序的一个优点是所述贝叶斯程序是模块化的,从某种意义上说,所述贝叶斯程序允许一个体添加任何可用的信息。在当前情况下,使用了片段大小信息,但是可以附加地或替代地包括其他特征,所述多个包括细胞游离胎儿DNA的其他特征,例如:多个优选的终止位置的簇。还设想了单倍型信息。富含细胞游离胎儿DNA的细胞游离DNA也可以通过根据本发明的一些实施例的所述方法进行处理。
本示例的所述分析中显示的所述准确度计算是基于多个未过滤的原始结果,因此,所述实际的准确度高于本说明书中的报告的准确度。所述准确度还取决于本研究中的未优化的多个功能。在此示例中,将预测应用于所有位点,即使是可信度较低的多个位点,并且返回在每个位点中使用的所有相关的信息。然后,可以应用多个不同的注释、多个统计测试及多个机器学习程序,以便过滤掉多个低可信度结果,如所述机器学习重新校准步骤中所示。所述贝叶斯程序与变异侦测的所述接受过程的一致性是所述贝叶斯程序允许的另一个优点,因为所述多个后验概率构成了用于过滤的所述校准参数。
本示例展示了外显子组测序及基因组测序。与深度WGS数据相比,当本多个实施例的所述方法应用于深度WES数据时,在所述母亲是杂合的多个位置处获得的所述准确度不高,所述多个结果是在怀孕的一早期的与临床相关的星期中获得,在所述星期中需要进行DNA扩增。通过无扩增WGS,本示例示出了即使在怀孕的所述早期阶段,所述方法也是准确的。为了在多个较小的基因组区域也达到高准确度,将会测试其他文库构建及多个测序方法。
本示例展示了可以利用例如NGS之类的可用技术获得大量SGD的所述NIPD,本示例中使用的所述概率标度确保了所述多个预测位点中的一些百分比具有一特定的预测。所述多个位点可用于下游分析,以发现尚未发现的多个稀有变异。由于WES的成本低,根据本发明的一些实施例,WES可用于大群体研究,由于使用WES可以创建一大型且统一的数据集,可以对所述数据集进行进一步分析以改善所述模型,因此,WES与常规技术相比具有优势。
示例2:
本示例示出了一深度学习程序可用于以一非侵入式方式准确检测所述胎儿中的多个小变异,在本发明的一些实施例中,所述深度学习程序可以代替示例1中使用的所述贝叶斯模型及所述机器学习重新校准过程,并且可以在一端到端框架中使用。所述多个实施例的优点中的一个是深度学习程序,所述深度学习程序可以自动对多个系统性错误建模。这在难以解释的数据(例如:低深度测序)中很有用。所述多个发明人发现所述端到端深度学习的性能优于所述贝叶斯模型。例如:所述深度学习程序可以处理低胎儿分数及测序覆盖率,所述低胎儿分数及所述测序覆盖率是本任务中的所述多个主要限制因素。
与贝叶斯模型不同,所述深度学习程序可以提供一未知的相互依赖的似然函数的一近似值,本功能利用了所有三个个体中覆盖一位置的多个读段以及多个相邻的核苷酸之间的所述关系。本发明的所述多个发明人发现深度学习程序的使用可以应用在利用多个长度以外的多个特征(例如:所述核苷酸序列本身)。所述深度学习程序可以应用于各种测序平台(因美纳,纳米孔(Nanopore)等)及多个方法(WES、WGS、面板等)。
图11总结了所述贝叶斯模型(在图11中称为霍巴里(Hoobari))及所述端到端深度学习模型(在图11中称为深度霍巴里(DeepHoobari))之间的所述多个差异。
人工神经网络:
所述人工神经网络可选地及优选地是一卷积神经网络(CNN)。
特征提取:
所述细胞游离DNA及所述父母的DNA的所述数据可以使用所述多个比对的读段及其元数据表示,例如:所述多个比对的读段的多个碱基质量(核苷酸水平的信息)或多个配对质量(读段水平的信息)或附件1的表A.1中列出的任何一个特征。替代地或附加地,可以使用变异侦测软件(例如但不限于:GATK的单倍型侦测(HaplotypeCaller)、谷歌(Google)的深度变异(DeepVariant)或免费贝叶斯Freebayes)提取与可能支持的多个等位基因有关的信息,所述信息是所述基因组基因座的所述水平,所述细胞游离DNA、母亲及父亲可以使用不同水平的数据。在本发明的一些实施例中,多个数据源被减少。例如:通过减少所使用的覆盖深度及细胞游离胎儿/或避免所述父母信息的使用。
细胞游离DNA的堆叠张量:
在所述学习阶段,与所述基因组中一特定的基因座相对应的所有数据可选地及优选地由一输入多维张量表示,所述输入多维张量与包含使用一侵入性测试所发现的所述真实的胎儿基因型的一标记相关联,所述CNN在每次迭代(向前及向后传播)中接收一批量的输入多维张量及所述批量的输入多维张量的多个标签。
所述张量可以是例如:覆盖以所述评估的SNP为中心围绕的一候选SNP的多个读数的一堆叠。在所述方式下,所述张量的前两个(例如:宽度及高度)对应于所述堆叠的所述长度及读段的所述次数,所述多个读段可以以任何技术来编码,包括但不限于:单热编码、组热编码等。所述张量的所述第三维度(例如:深度)是所述CNN的多个输入通道的所述数目,每个通道为所述SNP提供元数据。在本发明的一些实施例中,通过多个乘法函数处理由多个通道表示的信息,其中所述多个乘法函数的输出构成所述张量内的所述多个通道。
实验设计:
多个数据集:
下面的表3A及3B列出了本研究中使用的所述多个数据集,所述研究旨在防止所述预测模型过度拟合。分析了四个家族:G1至G2家族包含高质量数据,并且在一先前的研究中己进行了测序;G5家族包括了高质量的数据,由于G5家族是从第一妊娠期开始测序,因此具有生物学上的挑战性;E1家族也具有挑战性,由于E1家族是使用WES测序的,WES测序被证明具有更多错误。
表3A:
表3B:
a)Chan等人,2016年;b)Kitzman等人,2013年;1)多个常染色体上发现的多个双等位基因变异,父母及胎儿深度≥5、细胞游离DNA深度≥20的宽松过滤;2)按照一无PCR的文库构建方案进行全基因组测序(WGS);3)中位数,达到目标;4)孕龄。
第1阶段:
所述研究的所述第一阶段是基于数据集G1选择所述数据的所述重新表达、所述张量的所述结构及所述网络的基础架构,所述数据集分为一训练集(所述多个变异的80%)及一测试集(所述多个变异的20%)。所述训练进一步分为90%的训练及10%的多个验证集。由于所述多个变异的数量仍然很大,因此,在所述实验中的大部分都需要对所述多个变异进行进一步采样。一旦选择了令人满意的架构,就只能在所述测试集上对所述架构进行一次测试一,所述架构还在G2进行了测试,以呈现一最佳情况,其中多个家族之间的所述多个技术差异是最小的(在同一实验室使用所述相同的方法对多个家族进行测序)。
第2阶段:
多个数据集G1及G2合并到一数据集,所述数据集分为训练集、验证集及测试集,在第1阶段中选择的所述架构已经过训练及测试。此后,在数据集G5上测试了所述架构,以显示一更具挑战性的场景,其中用于训练的所述多个家族及用作测试的所述多个家族之间存在多个技术差异,这证明了所述模型的普遍性。
第3阶段:
数据集E1类似地划分,但具有一较大的测试集,以保持一足够数量的变异。所述先前阶段的所述模型用于转移学习,其中对所述训练后的模型进行了进一步的训练及微调。在一单独的或替代的过程中,仅基于E1的所述训练集来训练所述前几个阶段的所述架构。
其他实验:
可以使用多个插入与缺失重复第1至3阶段的所述多个实验,多个进一步的实验可以包括(1)对测序深度及胎儿分数进行下采样;(2)排除来自所述父亲的数据;(3)排除父母双方的数据;(4)将所需的父母的信息减少为仅基因型,使得只有所述细胞游离DNA才需要NGS。
多个结果:
在第1阶段中,所述张量基于多个比对的读段,并且GATK的单倍型侦测(HaplotypeCaller)用于执行一更准确的重新比对以及为每个读段分配每个读段的支持的等位基因。由于单倍型侦测(HaplotypeCaller)不是为多个怀孕的细胞游离DNA样本设计的并且会将所述多个样本视为来自一个个体,因此,在不侦测所述基因型的情况下分配多个等位基因的所述分配。发现基于所述细胞游离DNA及父母的所述多个读段比对的一张量与霍巴里(Hoobari)的所述准确度相匹配。所述多个结果示出在图12A至12P,对于所述训练集(图12A、12B、12E、12F、12I、12J、12M、12N)及所述验证集(图12C、12D、12G、12H、12K、12L、12O、12P)示出了简单的CNN(红色)、具有按片段长度排序的多个细胞游离DNA的读段的CNN(绿色)、具有按片段长度排序的细胞游离DNA读段的CNN及由支持的等位基因排序的父母读段(蓝色)以及初始阶段(Inception)第3版的CNN(黄色)。对于准确度在图12A至12P中示出,针对所述训练集(图12A);针对所述验证集(图12C);根据亲本基因型分为3组变异:仅有母源性杂合(mat-het)、仅有父源性杂合(pat-het)或父母两者均为杂合(两者),对于所述3种可能的胎儿基因型:与所述参考等位基因纯合(homref)、所述变异的等位基因(homalt)及所述杂合子(het)。所述缺失的所述多个结果(图12B,12D)在所述多个模型之间不具有比较性,因此不予考虑。
在两个父母都是杂合子的位置,以前很难预测所述胎儿的基因型,与霍巴里(Hoobari)相比,结果示出了一显著的改进。当通过支持的等位基因对所述多个亲本读段进行排序以及按片段长度对所述细胞游离DNA进行排序时,所述多个结果得到改进(每个读段都来自具有一不同长度的片段)。
应当理解,可以采用更复杂的张量及多个网络架构。例如:在一单热编码张量中,每个核苷酸由代表其碱基质量的1及0的一组合表示,再乘以所述各自的可信度。所述碱基A、C、G及T可以在四个通道上进行编码,例如:分别编码为1000、0100、0010及0001。例如:如果所述碱基A的可信度为0.999,则为第一个通道被分配1*0.999=0.999,而其他通道将显示0*0.999=0。另一个示例是一组热编码模型,其中所述热编码的核苷酸乘以所有其他特征的一向量,所述数据的所述多个表示的所述多个结果在图13A至13P中示出,多个表示为所述单热(蓝色)及组热(粉红色)。对于如图12A至12P所示的多个相同的群组示出了准确度及缺失。如图13A至13P所示,所述单热及组热表示提高了所述CNN至学习所述数据的所述多个特征的能力。
图14A至14P示出了使用所述父母信息仅包括所述父母的基因型而没有读段水平或核苷酸水平的信息的一模型所获得的多个结果,所述数据的所述多个表示的所述多个结果在图13A至13P中示出,多个表示为单热(蓝色)、组热(浅蓝色),并且对于仅有使用的父母信息是所述多个父母基因型(与所述参考或变异的等位基因纯合或杂合)的所述情况(粉红色),如图12A至12P所示的多个相同的群组示出了准确度及缺失。。
图14A至14P展示了即使仅使用所述多个亲本基因型也可以获得很高的准确度,这意味着没有必要使用NGS对所述多个父母进行测序,并且其他技术也可以满足要求。然而,来自NGS的读段及核苷酸水平的信息似乎可以改善所述多个结果。
附件1:
表A.1:
所述多个机器学习模型中使用的多个特征:
虽然本发明已经结合其特定实施例进行了描述,但是显而易见的是,许多备选方案,修饰以及变化对本领域技术人员来说是显而易见的。因此,本发明旨在涵盖所有落入所述权利要求的精神和范围内的所有这样的备选方案、修饰以及变化。
在本说明书中提及的所有出版物、专利及专利申请以其整体作为参考文献并入本说明书中,其程度如同各独立的出版物、专利或专利申请案被明确地且个别地标示为以引用的方式并入本文中。此外,本申请中任何参考文献的引用或证明不应被解释为承认所述参考文献可作为本发明的现有技术。本申请中标题部分在本文中用于使本说明书容易理解,而不应被解释为必要的限制。
另外,本申请的任何(多个)优先权文件在此以引用方式全部并入在本说明书中。
多个参考文献:
1.Lo、Y.M.D.等人,用于胎儿染色体非整倍性的所述分子检测的数字PCR,美国国家科学院学报104,13116至13121(2007)。
2.FanH.C.、BlumenfeldY.J.、Chitkara U.、HudginsL.及QuakeS.R.通过散弹枪对母体血液中DNA的测序对胎儿非整倍性进行非侵入性诊断,美国国家科学院学报105,16266-16271(2008)。
3.HillM.、ComptonC.、Lewis C.、SkirtonH.及ChittyL.S.,对有血友病风险的多个孕妇中胎儿性别的确定:探讨英国的多个医疗专业人员的所述多个临床实践及多个态度的一定性研究,血友病18,575至583(2012)。
4.LewisC.、HillM.、SkirtonH.及ChittyL.S.,用于确定胎儿性别的非侵入性产前诊断:从所述服务的使用者的角度的多个利与弊,欧洲人类遗传学20,1127至1133(2012)。
5.FinningK.、MartinP.及DanielsG.在英国的临床服务,使用母体血浆中的游离胎儿DNA预测胎儿恒河猴(Rh)D血型,纽约科学院年鉴1022,119至123(2004)。
6.MinonJ.-M.、GerardC.、SenterreJ.-M.、SchaapsJ.-P.及FoidartJ.-M.,以母体血浆进行常规胎儿RHD基因分型:在比利时的一个四年经验,输血(巴黎)48,373至381(2008年)。
7.MahdiehN.及RabbaniB.,多个遗传疾病中多个突变检测方法的一概述,伊朗儿科杂志23,375至388(2013)。
8.YangY.等人,临床全基因组测序对孟德尔疾病的诊断,新英格兰医学杂志369,1502至1515(2013)。
9.Isakov O.、Perrone M.及Shomron N.,外显子组测序分析:疾病变异检测的一指南,方法生物学,新泽西州克利夫顿1038,137至158(2013)。
10.MengL.等人,多个重症监护病房的多个婴儿的外显子组测序的应用:确定多个严重的单基因疾病及对医疗管理的影响,美国医学会小儿科期刊171,e173438至e173438(2017)。
11.Mackie手F.L.、CarssK.J.、HillmanS.C.、HurlesM.E.及KilbyM.D.具有多个结构性畸形的多个胎儿的外显子组测序,临床医学杂志3,747至762(2014)。
12.VoraN.L等人,多个异常胎儿的产前外显子组测序:多个新的机遇及挑战,医学遗传学19,1207(2017)。
13.KitzmanJ.O.等人,一人类胎儿的非侵入性全基因组测序,科学转化医学4,137ra76(2012)。
14.FanH.C.等人,所述胎儿基因组的非侵入性产前测量,自然487,320至324(2012)。
15.HillM.等人,用于囊性纤维化的非侵入性产前诊断:检测多个父系突变、探索多个患者喜好及成本分析,产前诊断35,950至958(2015)。
16.LunF.M.F.等人,通过数字大小选择及母体血浆中的DNA的相对突变剂量对多个单基因疾病进行非侵入性产前诊断,美国国家科学院学报105,19920至19925(2008)。
17.LamK.-WG.等人,母体血浆的靶向大规模平行测序对多个单基因疾病的非侵入性产前诊断:在β-地中海贫血中的应用,临床化学58,1467至1475(2012)。
18.LoY.M.D.等人,母体血浆DNA测序揭示了所述胎儿的所述全基因组遗传及突变特征,科学转化医学2,61ra91至61ra91(2010)。
19.ChenS.等人,通过母体血浆测序的由单倍型辅助的准确非侵入性胎儿全基因组的恢复,基因组医学5,18(2013)。
20.ChanK.C.A。等人,第二代非侵入性胎儿基因组分析揭示了新生突变、单碱基亲本遗传及多个优选的DNA末端,美国国家科学院学报201615800(2016),数字对象识别符(doi):10.1073/pnas.1615800113
21.SnyderM.W.等人,非侵入性胎儿基因组测序:一引物,产前诊断33,547至554(2013)。
22.SnyderM.W.、AdeyA.、KitzmanJ.O.及ShendureJ.,单倍型分辨的基因组测序:多个实验方法及多个应用,遗传学自然评论16,344至358(2015)。
23.JenkinsL.A.、DeansZ.C.、LewisC.及AllenS.为多个单基因疾病提供的一经认可非侵入性产前诊断服务,并且提供多个最佳实践建议,产前诊断(2017),doi:10.1002/pd.5197
24.ChanK.C.A.等人,母体血浆中母体及胎儿DNA的多个大小分布,临床化学50,88至92(2004)。
25.FanH.C.、BlumenfeldY.J.、Chitkara U.、HudginsL.及QuakeS.R.,通过双端测序分析胎儿及母体细胞游离DNA的大小分布,临床化学56,1279至1286(2010)。
26.YuS.C.Y.等人,使用血浆DNA进行非侵入性产前检测的多个基于大小的分子诊断,美国国家科学院学报111,8583至8588(2014)。
27.CiriglianoV.、
28.SunK。等人,咖啡:使用母体血浆DNA进行无控制的非侵入性胎儿染色体检查,产前诊断37,336至340(2017)。
29.SillenceK.富集用于非侵入性性产前检查(NIPT)的细胞游离胎儿DNA(cffDNA):多个分子技术的一比较(2016)。
30.MullaneyJ.M.、MillsR.E.、PittardW.S.及DevineS.E.的人类基因组中的多个小插入与缺失(INDEL),人类分子遗传学19,R131至R136(2010)。
31.NeumanJ.A.、IsakovO.及ShomronN.,从深度测序数据中的插入与缺失的分析:用于实现最佳检测的软件评估,生物信息学简介14,46至55(2013)。
32.JiangY.、TurinskyA.L.及BrudnoM.,所述缺失的插入与缺失:对一人类基因组中插入与缺失变异的一估计以及阻碍检测的多个因素分析,核酸研究43,7217至7228(2015)。
33.HwangS.、KimE.、LeeI.及MarcotteE.M.,使用多个金标准个人外显子组变异体的多个变异体侦测管道的系统比较,科学报告5,17875(2015)。
34.Li、H.及DurbinR.使用伯罗斯惠勒(Burrows-Wheeler)变换进行快速准确的短读比对,生物信息学25,1754至1760(2009)。
35.GarrisonE.及MarthG.,基于短读测序的基于单倍型的变异检测,ArXiv12073907Q-生物(Q-Bio)(2012)。
36.DanecekP.等人,所述变体侦测格式及多个VCF工具(VCFtool),生物信息学27,2156至2158(2011)。
37.序列比对/图谱格式规范,2018年5月22日,www.samtools.github.io/hts-specs/SAMv1.pdf。
38.Chan KCA、Jiang P、Sun K、Cheng YKY、Tong YK、Cheng SH、Wong AIC、Hudecova I、Leung TY、Chiu RWK等人,2016年,第二代非侵入性胎儿基因组分析揭示了新生突变、单碱基父母遗传及优选的多个DNA末端,美国国家科学院院刊201615800。
39.Fan HC、Gu W、Wang J、Blumenfeld YJ、El-Sayed YY、Quake SR.,2012,所述胎儿基因组的非侵入性产前测量日,自然487:320至324。
40.Kitzman JO、Snyder MW、Ventura M、Lewis AP、Qu R、Simmons LE、GammillHS、Rubens CE、Santillan DA、Murray JC等人,2012,一人类胎儿的非侵入性全基因组测序,科学转化医学4:137ra76。
41.Luo R、Sedlazeck FJ、Lam T-W、Schatz M.,2018年,千里眼(Clairvoyante):一种用于单分子测序中的变异侦测的一多任务卷积深度神经网络,bioRxiv310458。
42.Poplin R、Chang P-C、Alexander D、Schwartz S、Colthurst T、Ku A、Newburger D、Dijamco J、Nguyen N、Afshar PT等人,2018年,创建具有深度神经网络的一通用SNP及小型插入与缺失的变异侦测器,bioRxiv 092890。
43.Torracinta R、Campagne F.,2016年,以多个神经网络训练多个基因型侦测器,bioRxiv097469。
- 识别血液中基因异常的方法及系统
- 血液样本中粒子的识别方法、系统及血液细胞分析仪