一种制备5-甲基-N-取代吡咯烷酮类化合物的方法
文献发布时间:2023-06-19 18:37:28
技术领域:
本发明涉及有机物制备技术领域,具体涉及一种制备5-甲基-N-取代吡咯烷酮类化合物的方法。
背景技术:
5-甲基-N-取代吡咯烷酮类化合物是一类含内酰胺结构的五元含氮杂环化合物。由于其结构特性以及适中的极性,其能够在工业生产中作为优良的溶剂,润滑剂,表面活性剂等。另外,含氮杂环类结构在医药与材料领域是重要的结构单元,因此对该类化合物的研究具有重大意义。
目前5-甲基-N-取代吡咯烷酮类化合物主要是通过乙酰丙酸与伯胺在催化剂在还原条件下制备得到的。制备5-甲基-N-取代吡咯烷酮类化合物的催化剂有镍催化剂,铂催化剂等。W.L.Shilling通过硅酸镍催化剂在氢气条件下,将乙酰丙酸与胺类转化为5-甲基-N-取代吡咯烷酮产物(US Patent 32355562,1996)。Fu利用Ru络合物作为催化剂,甲酸作为还原剂,将乙酰丙酸与胺类转化为5-甲基-N-取代吡咯烷酮产物,产率为30%~81%不等(ChemSusChem 2011,4,1578-1581)。Han利用负载在二氧化钛的铂催化剂,实现了常温常压乙酰丙酸与胺类转化为5-甲基-N-取代吡咯烷酮的反应(J.Am.Chem.Soc.2019,141,4002-4009)。
现有技术公开的制备5-甲基-N-取代吡咯烷酮类化合物的反应条件苛刻,或贵金属载量较高,因此这些方法的实用性较低。
发明内容:
本发明解决了现有技术存在的问题,提供一种制备5-甲基-N-取代吡咯烷酮类化合物的方法,本发明提供的方法能够高效、温和的制备5-甲基-N-取代吡咯烷酮类化合物,使5-甲基-N-取代吡咯烷酮类化合物的产率较高,具有较好的实际应用价值。
本发明的目的是一种铂系金属催化剂的制备方法,包括以下步骤:
(1)将铁源与络合剂溶于水中得到含铁的水溶液,将含铁的水溶液滴入硅源和碱的混合物中进行水热反应,得到铂系金属催化剂的载体前驱体,将所述的载体前驱体进行干燥和第一焙烧,得到铂系金属催化剂的载体(Fe-S-1载体);
(2)将步骤(1)得到的铂系金属催化剂的载体与铂源在水中进行浸渍,得到铂系金属催化剂的前驱体,将所述的前驱体在还原条件下进行第二焙烧,得到铂系金属催化剂。含铁的水溶液中铁的浓度为0.25mol/L。
优选地,步骤(1)所述的铁源为溶于水的铁盐,硅源为原硅酸酯,络合剂为胺类或胺类衍生物,碱为烷基氢氧化铵,铁源中铁离子与络合剂的摩尔比为1:1,硅源和碱的摩尔比比1:1。
优选地,步骤(1)所述的铁源选自硝酸铁,醋酸铁和硫酸铁中的一种以上,硅源选自原硅酸甲酯,原硅酸乙酯和原硅酸丁酯中的一种以上,络合剂选自乙二胺,乙二胺四乙酸,乙二胺四乙酸二钠和N,N,N,N-四甲基乙二胺中的一种以上,碱选自四甲基氢氧化铵,四乙基氢氧化铵和四异丙基氢氧化铵中的一种以上。
优选地,步骤(1)所述的水热反应的温度为100℃~250℃,时间为36~144小时,第一焙烧的温度为400℃~700℃,焙烧时间为2~6小时。
优选地,步骤(2)所述的浸渍的时间为8~24小时,还原条件为氢气和氮气的混合气氛,混合气氛中氢气和氮气的体积比为1:5~10,第二焙烧的温度为100℃~400℃,焙烧时间为0.5~1.5小时。
本发明还保护上述制备方法得到的铂系金属催化剂,所述的铂系金属催化剂包括载体以及负载在载体上的活性组分,活性组分包括铂和铁,铂系金属催化剂中铂的含量为0.5wt%~10wt%,铁的含量为0.1wt%~5wt%,载体选自Fe-S-1、Fe-S-1-EDTA、气相二氧化硅、二氧化钛、活性炭、三氧化二铝和介孔碳中的一种。
优选地,所述的铂系金属催化剂中铂的含量为0.8wt%~5wt%,铁的含量为0.5wt%~2wt%。
本发明还保护一种制备5-甲基-N-取代吡咯烷酮类化合物的方法,包括以下步骤:在所述的铂系金属催化剂的作用下,将乙酰丙酸与伯胺或一级硝基化合物在溶剂中进行缩合-还原反应,得到5-甲基-N-取代吡咯烷酮类化合物。伯胺或一级硝基化合物具体优选为苯胺、一正丁胺或硝基苯。
优选地,所述的铂系金属催化剂、乙酰丙酸与伯胺或一级硝基化合物的质量比为1:(0.5~20):(0.5~20)。
优选地,所述的缩合-还原反应的反应温度为10℃~250℃,反应压力为0.1~6MPa,反应时间为2~5小时。
本发明与现有技术相比,具有如下优点:
1、本发明提供的方法采用以铂和铁为有效成分的铂系金属催化剂催化乙酰丙酸与伯胺或一级硝基化合物的缩合-还原反应,得到5-甲基-N-取代吡咯烷酮类化合物,铂组分具有较好的加氢能力,能够使乙酰丙酸与伯胺缩合得到的不饱和内酰胺中的C=C双键进行还原;铁成分具有较好的路易斯(Lewis)酸性与对氮原子的亲和性,能够有效吸附底物中的伯胺成分,并使得其易于与酸发生缩合反应;在铂和铁的双重作用下,能够将乙酰丙酸与伯胺或一级硝基化合物高效、高选择性的转化为5-甲基-N-取代吡咯烷酮类化合物,使5-甲基-N-取代吡咯烷酮类化合物的产率较高。
2、此外本发明提供的方法能够在很低的贵金属载量下(<1wt%),在常温常压条件下高效实现5-甲基-N-取代吡咯烷酮类化合物的制备,有效降低了5-甲基-N-取代吡咯烷酮类化合物的生产成本。采用本发明提供的方法制备5-甲基-N-取代吡咯烷酮类化合物,5-甲基-N-取代吡咯烷酮类化合物的产率最高为94%。
附图说明:
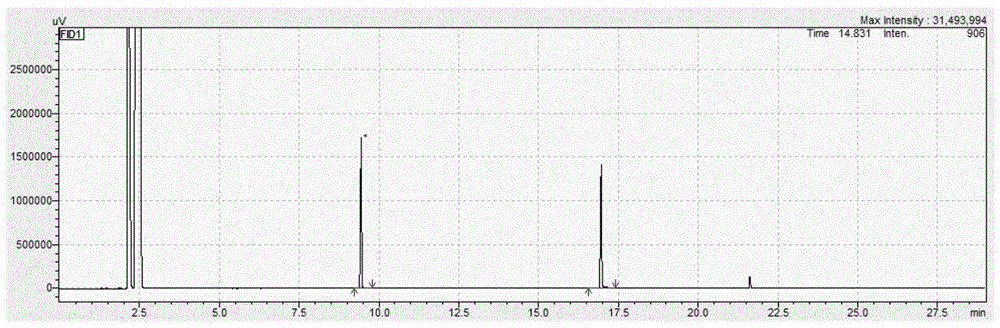
图1为本发明实施例8得到的反应产物的气相色谱图;
图2为本发明实施例8得到的反应产物的核磁共振氢谱图;
图3为本发明实施例26得到的反应产物的气相色谱图;
图4为本发明实施例26得到的反应产物的核磁共振氢谱图。
具体实施方式:
以下实施例是对本发明的进一步说明,而不是对本发明的限制。
除非另有定义,下文中所使用的所有专业术语与本领域技术人员通常理解含义相同。本文中所使用的专业术语只是为了描述具体实施例的目的,并不是旨在限制本发明的保护范围。除特别说明,本文中的实验材料和试剂均为本技术领域常规市购产品。
一种制备5-甲基-N-取代吡咯烷酮类化合物的方法,包括以下步骤:在铂系金属催化剂的作用下,将乙酰丙酸与伯胺或一级硝基化合物在溶剂中进行氢解反应(缩合-还原反应),得到5-甲基-N-取代吡咯烷酮类化合物。
铂系金属催化剂为负载型双金属催化剂,铂系金属催化剂的有效活性成分包括铂和铁。
本发明优选将铂系催化剂,乙酰丙酸与伯胺或一级硝基化合物混合,得到混合产物,再向混合产物中加入溶剂,将得到的混合溶液进行氢解反应(缩合-还原反应),得到N-取代吡咯烷酮类化合物。在本发明中,将铂系催化剂,乙酰丙酸与伯胺或一级硝基化合物混合的温度优选为15℃~30℃,更优选为18℃~26℃,最优选为20℃~25℃;将铂系催化剂乙酰丙酸,伯胺或一级硝基化合物混合的压力优选为0.8×10
在本发明中,铂系金属催化剂的有效活性成分包括铂和铁,优选包括铂单质和氧化铁。铂系金属催化剂中铂的含量优选为0.5wt%~10wt%,更优选为0.75wt%~5wt%,进一步优选为1wt%;铂系金属催化剂中铁的含量优选为0.1wt%~5wt%,更优选为0.5wt%~2wt%,进一步优选为0.8wt%~1.2wt%,最优选为1wt%。
在本发明中,铂系金属催化剂为负载型双金属催化剂,铂系金属催化剂还包括载体。在本发明中,铂系金属催化剂的载体优选为自制的Fe-S-1-EDTA载体、气相二氧化硅、二氧化钛(TiO
本发明对自制的Fe-S-1-EDTA载体的来源没有特殊的限制,可采用本领域技术人员熟知的制备硅基分子筛的方法制备得到。
本发明对铂系金属催化剂的来源没有特殊的限制,可采用本领域技术人员熟知的制备铂系金属催化剂的方法制备得到;在本发明中,铂系金属催化剂优选按照下述方法制备得到:铂系金属催化剂优选按照下述方法制备得到:将铁源,硅源,络合剂与碱在溶剂中进行水热,得到载体前驱体,将载体前驱体进行干燥和第一焙烧,得到铂系金属催化剂的载体;将铂系金属催化剂的载体与铂源在溶剂中进行浸渍,得到铂系金属催化剂的前驱体;将铂系金属催化剂的前驱体在还原条件下进行第二焙烧,得到铂系金属催化剂。
本发明对铁源,硅源,络合剂与碱的混合方法没有特殊的限制,采用本领域技术人员熟知的混合的技术方案,将上述铁源,硅源,络合剂与碱混合均匀即可。本发明优选在干燥的条件下进行混合。在本发明中,混合的时间优选为8~24小时,更优选为10~16小时,最优选为11~13小时。
本发明优选在水热的条件下进行载体前驱体合成,本发明对水热的方法没有特殊的限制,采用本领域技术人员熟知的搅拌的技术方案即可。本发明优选采用本领域技术人员熟知的具四氟乙烯内衬的不锈钢水热釜进行水热即可,如可从市场购买获得。在本发明中,水热的温度优选为100℃~250℃,更优选为150℃~200℃,最优选为170℃;水热的时间优选为36~144小时,更优选为72~96小时,最优选为84小时。
本发明得到载体前驱体后,将载体前驱体进行干燥和第一焙烧,得到载体Fe-S-1。在本发明中,干燥的方法优选为烘干,干燥的设备优选为烘箱。在本发明中,干燥的温度优选为70℃~100℃,更优选为75℃~90℃,最优选为80℃。
将载体前驱体干燥后,本发明将得到的干燥产物进行第一焙烧,得到载体Fe-S-1。在本发明中,第一焙烧的焙烧温度优选为400℃~700℃,更优选为500℃~600℃,最优选为550℃;第一焙烧的焙烧时间优选≥2小时,更优选为2~6小时,进一步优选为3~5小时,最优选为4小时。
本发明优选将铂源和载体按比例称量后混合,将得到的混合物加入到反应容器中在溶剂中进行浸渍,得到浸渍产物;本发明对浸渍用溶剂的种类没有特殊的限制,能够为所述铂源和载体提供浸渍环境即可。本发明对浸渍用溶剂的加入量没有特殊的限制,所加入的溶剂能浸没反应容器中的载体即可。在本发明中,浸渍用溶剂优选为水,更优选为蒸馏水或去离子水。
本发明对铂源和载体的混合方法没有特殊的限制,采用本领域技术人员熟知的混合的技术方案,将上述铂源、铁源和载体混合均匀即可。本发明优选在干燥的条件下进行混合。在本发明中,浸渍的时间优选为8~24小时,更优选为10~16小时,最优选为11~13小时。
本发明优选在搅拌的条件下进行浸渍,本发明对搅拌的方法没有特殊的限制,采用本领域技术人员熟知的搅拌的技术方案即可。本发明优选采用搅拌磁子进行搅拌。本发明对搅拌磁子的种类和来源没有特殊的限制,采用本领域技术人员熟知的搅拌磁子即可,如可由市场购买获得。
在本发明中,铂源优选为铂盐或铂酸,更优选为氯化铂(II)、六氯合铂(IV)酸和乙酰丙酮铂(II)中的一种或几种,最优选为六氯合铂(IV)酸;铁源优选为铁盐,更优选为硝酸铁,醋酸铁,硫酸铁中的一种或两种,最优选为硝酸铁;硅源优选为原硅酸酯,更优选为原硅酸甲酯,原硅酸乙酯,原硅酸丁酯中的一种或几种,最优选为原硅酸乙酯;络合剂优选为胺类或胺类衍生物,更优选为乙二胺,乙二胺四乙酸,乙二胺四乙酸二钠,N,N,N,N-四甲基乙二胺中的一种或几种,最优选为乙二胺四乙酸二钠;碱优选为烷基氢氧化铵,更优选为四甲基氢氧化铵,四乙基氢氧化铵,四异丙基氢氧化铵中的一种或几种,最优选为四乙基氢氧化铵;载体与上述技术方案所述的载体一致,在此不再赘述。本发明对铂源和载体的比例没有特殊的要求,比例使铂系金属催化剂中铂的含量与铁的含量满足上述技术方案中所述的铂系金属催化剂中铂的含量和铁的含量即可。本发明对铂源、铁源、硅源、络合剂和碱的来源没有特殊的限制,可以由市场购买获得,也可以采用本领域技术人员熟知的制备方法制备得到上述铂源、铁源,硅源,络合剂和碱。
得到浸渍产物后,本发明将所述浸渍产物进行干燥,得到铂系金属催化剂的前驱体。在本发明中,干燥的方法优选为烘干,干燥的设备优选为烘箱。在本发明中,所述干燥的温度优选为70℃~100℃,更优选为75℃~90℃,最优选为80℃。
将浸渍产物干燥后,本发明将得到的铂系金属催化剂的前驱体在还原条件下进行第二焙烧,得到铂系金属催化剂。在本发明中,还原条件优选为氢气和氮气的还原条件,混合气氛中氢气和氮气的体积比为1:5~10。本发明可以在静止的还原气氛下进行第二焙烧,也可以在流动的还原气氛下进行第二焙烧。在本发明中,当还原条件为流动的氢气和氮气时,氢气的流速优选为5~15mL/min,更优选为7~12mL/min,最优选为10mL/min;氮气的流速优选为50~150mL/min,更优选为70~120mL/min,最优选为90mL/min。
在本发明中,第二焙烧的加热方法优选为程序升温加热;程序升温加热为:以第一升温速率加热,温度从第一温度升至第二温度,第一温度优选为15℃~25℃,更优选为18℃~22℃,最优选为20℃;第二温度优选为100℃~400℃,更优选为200℃~300℃,最优选为250℃。
在本发明中,第一升温速率优选为9~11℃/min,更优选为9.5~10.5℃/min,最优选为10℃/min。在本发明中,恒温的时间优选为0.5~1.5小时,更优选为0.8~1.2小时,最优选为1小时。
本发明对乙酰丙酸,伯胺或一级硝基化合物的来源没有特殊的限制,可以由市场购买获得,也可以采用本领域技术人员熟知的制备乙酰丙酸,伯胺或一级硝基化合物的方法制备得到。
在本发明中,铂系金属催化剂,乙酰丙酸和伯胺或一级硝基化合物的质量比优选为1:(0.5~20):(0.5~20),更优选为1:(1~15):(1~15),进一步优选为1:(2.5~12):(2.5~12),最优选为1:(5~10):(5~10)。本发明对制备5-甲基-N-取代吡咯烷酮类化合物所用溶剂的种类和用量没有特殊的限制,采用本领域技术人员熟知的可用于制备5-甲基-N-取代吡咯烷酮类化合物的溶剂及用量即可;在本发明中,用于制备5-甲基-N-取代吡咯烷酮类化合物的溶剂优选为甲醇;铂系金属催化剂和用于制备5-甲基-N-取代吡咯烷酮类化合物的溶剂的质量比优选为1:(25~10000),更优选为1:(50~1000),进一步优选为1:(100~300),最优选为1:(150~250)。
本发明将上述铂系金属催化剂,乙酰丙酸,伯胺或一级硝基化合物,铂系金属催化剂和溶剂混合,得到混合溶液后,优选向上述混合溶液中充入氢气,进行氢解反应,得到5-甲基-N-取代吡咯烷酮类化合物。下述实施例中,5-甲基-N-取代吡咯烷酮类化合物优选为5-甲基-N-苯基吡咯烷酮或5-甲基-N-丁基吡咯烷酮。
本发明优选在干燥的条件下进行氢解反应,得到5-甲基-N-取代吡咯烷酮类化合物。本发明优选在搅拌的条件下进行氢解反应,得到5-甲基-N-取代吡咯烷酮类化合物。本发明优选采用搅拌磁子进行搅拌,本发明对搅拌磁子的种类和来源没有特殊的限制,采用本领域技术人员熟知的搅拌磁子即可,如可由市场购买获得。本发明优选在密封的条件下进行氢解反应,得到5-甲基-N-取代吡咯烷酮类化合物;本发明对密封条件的获得,没有特殊的限制,优选在密封仪器中进行氢解反应,得到5-甲基-N-取代吡咯烷酮类化合物。在本发明中,密封仪器优选为带磨口的常压反应管,更优选为带有搅拌磁子的带磨口的常压反应管,最优选为干燥的带有搅拌磁子的带磨口的常压反应管。
本发明优选排出密封仪器中的空气后,再将混合溶液进行氢解反应,得到5-甲基-N-取代吡咯烷酮类化合物。本发明对排出密封仪器中空气的方法没有特殊限制,采用本领域技术人员熟知的排出密封仪器中空气的技术方案即可。本发明优选采用向密封仪器中充入氢气再放气的方法排出密封仪器中的空气。在本发明中,放气的次数优选为4~6次,更优选为5次。
将密封仪器中的空气排出后,本发明向密封仪器中充入氢气,本发明优选在充入氢气后检查所述密封仪器的密封性,然后进行氢解反应(缩合还原反应),得到5-甲基-N-取代吡咯烷酮类化合物。本发明对所述检查密封仪器密封性的方法没有特殊的限制,采用本领域技术人员熟知的检查仪器密封性的技术方案即可。
本发明可以按照下述步骤检查密封仪器的密封性:向密封仪器中充入氢气后,将密封仪器密封,然后将密封仪器浸没于水中,观察所述密封仪器各处是否有气泡逸出,若无气泡逸出,则说明密封仪器的密封性良好。
在本发明中,氢解反应的反应温度优选为0℃~250℃,更优选为20℃~100℃,最优选为30℃~50℃;氢解反应的反应压力优选为0.1~6MPa,更优选为0.15~3MPa,最优选为0.2MPa;氢解反应的反应时间优选为2~5小时,更优选为2.5~4小时,最优选为3小时。
氢解反应完成后,本发明将得到的反应溶液中的铂系金属催化剂、溶剂和反应产物进行分离,回收得到的铂系催化剂和溶剂可以重复利用。本发明优选将得到的反应溶液采用减压蒸馏的方法进行溶剂和反应产物的分离,分别收集反应溶液中的溶剂和N-取代吡咯烷酮类化合物。本发明对减压蒸馏的方法没有特殊的限制,采用本领域技术人员熟知的减压蒸馏的技术方案即可。在本发明中,反应溶液中溶剂的收集温度优选为40℃~70℃,更优选为42℃~50℃,最优选为45℃。反应溶液中的收集压力优选为0.005~0.05MPa,更优选为0.005~0.02MPa,最优选为0.01MPa。
本发明采用铂系金属催化剂催化乙酰丙酸与伯胺或一级硝基化合物制备5-甲基-N-取代吡咯烷酮类化合物,能够使乙酰丙酸与伯胺在单一反应溶剂中直接反应,得到5-甲基-N-取代吡咯烷酮类化合物,避免了使用双相体系反应溶剂或离子液体反应溶剂,使制备得到的5-甲基-N-取代吡咯烷酮类化合物易于分离。
缩合还原反应完成后,本发明优选将得到的反应溶液采用离心的方法回收所述反应溶液中的铂系金属催化剂。本发明对回收反应溶液中的铂系金属催化剂、溶剂和5-甲基-N-取代吡咯烷酮类化合物的时间顺序没有特殊的限制,可以在回收得到反应溶液中的溶剂和5-甲基-N-取代吡咯烷酮类化合物之前回收铂系金属催化剂,也可以在回收得到所述反应溶液中的溶剂和5-甲基-N-取代吡咯烷酮类化合物之后回收铂系金属催化剂。本发明将回收得到的铂系金属催化剂在溶剂中冲洗3~4次即可再次用于制备5-甲基-N-取代吡咯烷酮类化合物。本发明对离心的方法没有特殊的限制,采用本领域技术人员熟知的离心的技术方案即可。在本发明中,冲洗铂系金属催化剂的溶剂优选为甲醇。本发明中的铂系金属催化剂可以重复使用,因此本发明提供的制备5-甲基-N-取代吡咯烷酮类化合物的方法易于进行大规模工业生产。
制备得到5-甲基-N-取代吡咯烷酮类化合物后,本发明采用气相色谱内标法检测了5-甲基-N-取代吡咯烷酮类化合物的产率。在本发明中,内标法为:在分析样品中某组分的含量时,加入一种内标物以校准和消除由于操作条件的波动而对分析结果产生的影响,加入的内标物可以被色谱柱所分离,而且不受样品中其他组分的干扰,只要测定内标物和待测组分的峰面积和相对响应值,即可得到待测组分在样品中的百分含量。
本发明采用内标法检测5-甲基-N-取代吡咯烷酮类化合物的产率,检测结果准确度高。在本发明中,内标法检测的内标物为苯甲醚。检测结果表明,采用本发明提供的方法制备5-甲基-N-取代吡咯烷酮类化合物,其产率最高为94%。
实施例1
一种铂系金属催化剂的制备方法,包括如下步骤:
将摩尔比为1:1的硝酸铁与乙二胺四乙酸二钠溶于一定量的水,配制成溶液,随后将该溶液滴入原硅酸四乙酯与四乙基氢氧化铵的混合物中充分混合,四乙基氢氧化铵与原硅酸四乙酯的摩尔比为1:1,使得到的混合物中,以Fe
将上述水热釜中的混合物在烘箱中于170℃下水热84小时,然后将得到的产物离心,水洗,并置于80℃的烘干箱中烘干,将得到的烘干产物在550℃下进行第一焙烧4小时,得到Fe-S-1载体。
将上述得到的Fe-S-1载体加入干燥的带有搅拌磁子的烧瓶中,加入六氯合铂(IV)酸,使得到的六氯合铂(IV)酸与Fe-S-1载体的混合物中,Pt的含量为1wt%。随后加入一定量的蒸馏水,使得液面恰好没过Fe-S-1载体。
将上述烧瓶中的六氯合铂(IV)酸和Fe-S-1载体的混合物浸渍12小时,然后将得到的浸渍产物置于80℃的烘干箱中烘干,得到以Fe-S-1为载体的铂催化剂的前驱体。
将上述得到的铂催化剂的前驱体在氢气流速为10mL/min,氮气流速为90mL/min的还原条件下进行第二焙烧,第二焙烧的过程为:以10℃/min的升温速率加热,使第二焙烧的焙烧温度从20℃升至250℃;然后保温1小时。
将上述采用自然降温的方法冷却到20℃后,得到以硅基分子筛为载体的铂-氧化铁催化剂。
实施例2
与实施例1相同,不同之处在于:调整六氯合铂(IV)酸和Fe-S-1载体的混合比例,使得到的六氯合铂(IV)酸与Fe-S-1载体的混合物中铂的含量为0.5wt%。
实施例3
与实施例1相同,不同之处在于:调整六氯合铂(IV)酸和Fe-S-1载体的混合比例,使得到的六氯合铂(IV)酸与Fe-S-1载体的混合物中铂的含量为5wt%。
实施例4
与实施例1相同,不同之处在于:调整硝酸铁与原硅酸四乙酯的混合比例,使得到的Fe-S-1载体中,以Fe
实施例5
与实施例1相同,不同之处在于:调整硝酸铁与原硅酸四乙酯的混合比例,使得到的Fe-S-1载体中,以Fe
实施例6
将十六烷基三甲基溴化铵与三乙醇胺溶液溶于水中,配制十六烷基三甲基溴化铵的浓度为100g/L,三乙醇胺的浓度为3.3g/L的混合溶液。对上述混合溶液超声处理2小时使之尽可能溶解,配制成溶液;随后将溶液转移至带搅拌子的容器中,于60℃下向将向该溶液中滴入环己烷,缓慢搅拌1h,随后向混合物中滴加正硅酸乙酯。将所述混合物在室温下搅拌24小时,离心收集固体产物,并于室温下风干,将得到的风干产物于550℃下第一焙烧4小时,得到球状硅胶载体。
将上述得到的球状硅胶载体加入干燥的带有搅拌磁子的烧瓶中,加入六氯合铂(IV)酸,使得到的六氯合铂(IV)酸与球状硅胶载体的混合物中,Pt的含量为1wt%。随后加入一定量的蒸馏水,使得液面恰好没过球状硅胶载体。
将上述烧瓶中的六氯合铂(IV)酸和球状硅胶载体的混合物浸渍12小时,然后将得到的浸渍产物置于80℃的烘干箱中烘干,得到以球状硅胶为载体的铂催化剂的前驱体。
将上述得到的铂催化剂的前驱体在氢气流速为10mL/min,氮气流速为90mL/min的还原条件下进行第二焙烧,第二焙烧的过程为:以10℃/min的升温速率加热,使第二焙烧的焙烧温度从20℃升至250℃;然后保温1小时。
将上述采用自然降温的方法冷却到20℃后,得到以球状硅胶为载体的铂催化剂。
实施例7
将一定比例的六氯合铂(IV)酸和气态二氧化硅称量后混合,使得到的混合物中铂的含量为1wt%,将所述混合物在带有搅拌磁子的干燥烧瓶中混合均匀,然后向所述烧瓶中加入恰好能浸没活性炭的蒸馏水。
将上述烧瓶中的六氯合铂(IV)酸和二氧化硅浸渍12小时,然后将得到的浸渍产物置于80℃的烘干箱中烘干,将得到的烘干产物在氢气流速为10mL/min,氮气流速为90mL/min的还原条件下进行焙烧,所述第二焙烧的过程为:以10℃/min的升温速率加热,使所述焙烧的焙烧温度从20℃升至250℃;然后保温1小时。
将上述焙烧后得到的产物采用自然降温的方法冷却到20℃后,得到以气态二氧化硅为载体的铂催化剂。
实施例8
将1mmol乙酰丙酸,1mmol一正丁胺和10mg实施例1制备的硅基分子筛为载体的铂-氧化铁催化剂在20℃、1标准大气压下加入到干燥的带有搅拌磁子的带磨口的常压反应管中,然后向所述高压反应釜中加入2mL的甲醇。
将上述反应管密封后充入氢气再放气,排出高压反应釜中的空气,放气的次数为6次,然后向反应管中充入氢气至0.1MPa后停止充气,检查反应管的密封性:将密封仪器浸没于水中,观察密封仪器各处是否有气泡逸出,若无气泡逸出,则说明密封仪器的密封性良好。
将反应管的物质在25℃下进行缩合-还原反应3小时,得到反应产物。
将上述得到的反应产物进行气相色谱测试和核磁共振氢谱测试,测试结果如图1,图2所示,图1为本发明实施例8得到的反应产物的气相色谱图;图2为本发明实施例8得到的反应产物的核磁共振氢谱谱图。测试结果表明,实施例8得到的反应产物为5-甲基-N-丁基吡咯烷酮。
将上述得到的5-甲基-N-丁基吡咯烷酮加入苯甲醚,然后对得到的混合物进行气相质谱检测,得到所述混合物的气相色谱图;计算气相色谱图中苯甲醚和5-甲基-N-丁基吡咯烷酮的峰面积和相对响应值,根据苯甲醚的加入量以及苯甲醚和5-甲基-N-丁基吡咯烷酮的峰面积和相对响应值计算得到上述混合物中5-甲基-N-丁基吡咯烷酮的含量,再根据5-甲基-N-丁基吡咯烷酮含量计算5-甲基-N-丁基吡咯烷酮的产率。检测结果为:实施例8得到的5-甲基-N-丁基吡咯烷酮的产率为94%。
实施例9
按照实施例8的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是采用80℃的反应温度替换实施例8中25℃的反应温度。
按照实施例8的检测方法检测实施例9得到的5-甲基-N-丁基吡咯烷酮的产率,检测结果为,实施例9得到的5-甲基-N-丁基吡咯烷酮的产率为81%。
实施例10
按照实施例8的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是采用0℃的反应温度替换实施例8中25℃的反应温度。
按照实施例8的检测方法检测实施例10得到的5-甲基-N-丁基吡咯烷酮的产率,检测结果为,实施例10得到的5-甲基-N-丁基吡咯烷酮的产率为63%。
实施例11
按照实施例8的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是采用1MPa的反应压力替换实施例8中0.1MPa的反应压力。
按照实施例8的检测方法检测实施例11得到的5-甲基-N-丁基吡咯烷酮的产率,检测结果为,实施例11得到的5-甲基-N-丁基吡咯烷酮的产率为92%。
实施例12
按照实施例8的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是采用乙醇作为反应溶剂替换实施例8中甲醇作为反应溶剂。
按照实施例8的检测方法检测实施例12得到的5-甲基-N-丁基吡咯烷酮的产率,检测结果为,实施例12得到的5-甲基-N-丁基吡咯烷酮的产率为90%。
实施例13
按照实施例8的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是采用异丙醇作为反应溶剂替换实施例8中甲醇作为反应溶剂。
按照实施例8的检测方法检测实施例13得到的5-甲基-N-丁基吡咯烷酮的产率,检测结果为,实施例13得到的5-甲基-N-丁基吡咯烷酮的产率为79%。
实施例14
按照实施例8的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是采用水作为反应溶剂替换实施例8中甲醇作为反应溶剂。
按照实施例8的检测方法检测实施例14得到的5-甲基-N-丁基吡咯烷酮的产率,检测结果为,实施例14得到的5-甲基-N-丁基吡咯烷酮的产率为9%。
实施例15
按照实施例8的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是采用四氢呋喃作为反应溶剂替换实施例8中甲醇作为反应溶剂。
按照实施例8的检测方法检测实施例15得到的5-甲基-N-丁基吡咯烷酮的产率,检测结果为,实施例15得到的5-甲基-N-丁基吡咯烷酮的产率为35%。
实施例16
按照实施例8的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是采用乙酸乙酯作为反应溶剂替换实施例8中甲醇作为反应溶剂。
按照实施例8的检测方法检测实施例16得到的5-甲基-N-丁基吡咯烷酮的产率,检测结果为,实施例16得到的5-甲基-N-丁基吡咯烷酮的产率为10%。
实施例17
按照实施例8的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是采用甲苯作为反应溶剂替换实施例8中甲醇作为反应溶剂。
按照实施例8的检测方法检测实施例17得到的5-甲基-N-丁基吡咯烷酮的产率,检测结果为,实施例17得到的5-甲基-N-丁基吡咯烷酮的产率为41%。
实施例18
按照实施例8的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是采用正己烷作为反应溶剂替换实施例8中甲醇作为反应溶剂。
按照实施例8的检测方法检测实施例18得到的5-甲基-N-丁基吡咯烷酮的产率,检测结果为,本发明实施例18得到的5-甲基-N-丁基吡咯烷酮的产率为14%。
实施例19
按照实施例8的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是采用γ-戊内酯作为反应溶剂替换实施例8中甲醇作为反应溶剂。
按照实施例8的检测方法检测实施例19得到的5-甲基-N-丁基吡咯烷酮的产率,检测结果为,实施例19得到的5-甲基-N-丁基吡咯烷酮的产率为20%。
实施例20
按照实施例8的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是采用实施例2中制备以硅基分子筛为载体的铂-氧化铁催化剂替换实施例8中实施例1中制备以硅基分子筛为载体的铂-氧化铁催化剂。
按照实施例8的检测方法检测实施例20得到的5-甲基-N-丁基吡咯烷酮的产率,检测结果为,实施例20得到的5-甲基-N-丁基吡咯烷酮的产率为38%。
实施例21
按照实施例8的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是采用实施例3中制备以硅基分子筛为载体的铂-氧化铁催化剂替换实施例8中实施例1中制备以硅基分子筛为载体的铂-氧化铁催化剂。
按照实施例8的检测方法检测实施例21得到的5-甲基-N-丁基吡咯烷酮的产率,检测结果为,实施例21得到的5-甲基-N-丁基吡咯烷酮的产率为93%。
实施例22
按照实施例8的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是采用实施例4中制备以硅基分子筛为载体的铂-氧化铁催化剂替换实施例8中实施例1中制备以硅基分子筛为载体的铂-氧化铁催化剂。
按照实施例8的检测方法检测实施例22得到的5-甲基-N-丁基吡咯烷酮的产率,检测结果为,实施例22得到的5-甲基-N-丁基吡咯烷酮的产率为85%。
实施例23
按照实施例8的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是采用实施例5中制备以硅基分子筛为载体的铂-氧化铁催化剂替换实施例8中实施例1中制备以硅基分子筛为载体的铂-氧化铁催化剂。
按照实施例8的检测方法检测实施例23得到的5-甲基-N-丁基吡咯烷酮的产率,检测结果为,实施例23得到的5-甲基-N-丁基吡咯烷酮的产率为77%。
实施例24
按照实施例8的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是采用实施例6中制备以球状硅胶为载体的铂催化剂替换实施例8中实施例1中制备以硅基分子筛为载体的铂-氧化铁催化剂。
按照实施例8的检测方法检测实施例24得到的5-甲基-N-丁基吡咯烷酮的产率,检测结果为,实施例24得到的5-甲基-N-丁基吡咯烷酮的产率为73%。
实施例25
按照实施例8的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是采用实施例7中制备以气相二氧化硅为载体的铂催化剂替换实施例8中实施例1中制备以硅基分子筛为载体的铂-氧化铁催化剂。
按照实施例8的检测方法检测实施例25得到的5-甲基-N-丁基吡咯烷酮的产率,检测结果为,实施例25得到的5-甲基-N-丁基吡咯烷酮的产率为71%。
实施例26
将1mmol乙酰丙酸,1mmol苯胺和10mg实施例1制备的硅基分子筛为载体的铂-氧化铁催化剂在20℃、1标准大气压下加入到干燥的带有搅拌磁子的带磨口的常压反应管中,然后向所述高压反应釜中加入2mL的甲醇。
将上述反应管密封后充入氢气再放气,排出高压反应釜中的空气,放气的次数为6次,然后向反应管中充入氢气至0.1MPa后停止充气,检查所述反应管的密封性:将密封仪器浸没于水中,观察密封仪器各处是否有气泡逸出,若无气泡逸出,则说明密封仪器的密封性良好。将反应管的物质在25℃下进行缩合-还原反应3小时,得到反应产物。
将上述得到的反应产物进行核磁共振氢谱测试和气相色谱测试,测试结果如图3,图4所示,图3为实施例26得到的反应产物的气相色谱谱图;图4为实施例26得到的反应产物的核磁共振氢谱谱图。测试结果表明,实施例26得到的反应产物为5-甲基-N-苯基吡咯烷酮。
将上述得到的5-甲基-N-苯基吡咯烷酮加入苯甲醚,然后对得到的混合物进行气相质谱检测,得到所述混合物的气相色谱图;计算气相色谱图中苯甲醚和5-甲基-N-苯基吡咯烷酮的峰面积和相对响应值,根据苯甲醚的加入量以及苯甲醚和5-甲基-N-苯基吡咯烷酮的峰面积和相对响应值计算得到上述混合物中5-甲基-N-苯基吡咯烷酮的含量,再根据5-甲基-N-苯基吡咯烷酮含量计算5-甲基-N-苯基吡咯烷酮的产率。检测结果为,实施例26得到的5-甲基-N-苯基吡咯烷酮的产率为71%。
实施例27
按照实施例26的技术方案制备得到5-甲基-N-苯基吡咯烷酮;不同的是采用硝基苯替换实施例26中的苯胺。
按照实施例26的检测方法检测实施例27得到的5-甲基-N-苯基吡咯烷酮的产率,检测结果为,实施例27得到的5-甲基-N-苯基吡咯烷酮的产率为31%。
实施例28
按照实施例26的技术方案制备得到5-甲基-N-苯基吡咯烷酮;不同的是采用5小时反应时间替换实施例26中的3小时反应时间。
按照实施例26的检测方法检测实施例28得到的5-甲基-N-苯基吡咯烷酮的产率,检测结果为,实施例28得到的5-甲基-N-苯基吡咯烷酮的产率为75%。
实施例29
与实施例1相同,不同之处在于:水热反应的温度为100℃,时间为144小时,第一焙烧的温度为400℃,焙烧时间为6小时,浸渍的时间为8小时,第二焙烧的温度为100℃,焙烧时间为1.5小时,得到以硅基分子筛为载体的铂-氧化铁催化剂。
将上述得到的以硅基分子筛为载体的铂-氧化铁催化剂按照实施例8的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是铂系金属催化剂、乙酰丙酸与伯胺或一级硝基化合物的质量比为1:0.5:0.5,缩合-还原反应的反应温度为10℃,反应压力为6MPa,反应时间为5小时。
实施例30
与实施例1相同,不同之处在于:水热反应的温度为250℃,时间为36小时,第一焙烧的温度为700℃,焙烧时间为2小时,浸渍的时间为24小时,第二焙烧的温度为400℃,焙烧时间为0.5小时,得到以硅基分子筛为载体的铂-氧化铁催化剂。
将上述得到的以硅基分子筛为载体的铂-氧化铁催化剂按照实施例8的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是所述的铂系金属催化剂、乙酰丙酸与伯胺或一级硝基化合物的质量比为1:20:20,缩合-还原反应的反应温度为250℃,反应压力为0.1MPa,反应时间为2小时。
对比例1
将一定比例的六氯合铂(IV)酸和二氧化硅称量后混合,使得到的混合物中铂的含量为1wt%,将混合物在带有搅拌磁子的干燥烧瓶中混合均匀,然后向烧瓶中加入恰好能浸没活性炭的蒸馏水。
将上述烧瓶中的六氯合铂(IV)酸和二氧化硅浸渍12小时,然后将得到的浸渍产物置于80℃的烘干箱中烘干,将得到的烘干产物在氢气流速为10mL/min,氮气流速为90mL/min的还原条件下进行焙烧,第二焙烧的过程为:以10℃/min的升温速率加热,使所述焙烧的焙烧温度从20℃升至250℃;然后保温1小时。
将上述焙烧后得到的产物采用自然降温的方法冷却到20℃后,得到以二氧化硅为载体的铂催化剂。
对比例2
按照对比例1的方法制备得到催化剂,不同的是以活性炭替换对比例1中的二氧化硅,得到以活性炭为载体的铂催化剂;催化剂中铂的含量为1wt%。
对比例3
按照对比例1的方法制备得到催化剂,不同的是以二氧化钛(TiO
对比例4
按照对比例1的方法制备得到催化剂,不同的是以三氧化二铝(Al
对比例5
按照对比例1的方法制备得到催化剂,不同的是以HZSM-5分子筛替换对比例1中的二氧化硅,得到以HZSM-5分子筛为载体的铂催化剂;催化剂中铂的含量为1wt%。
对比例6
按照实施例6的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是采用实施例1中制备的以Fe-S-1载体替换实施例6中实施例1制备的以Fe-S-1为载体的铂催化剂。
按照实施例8的检测方法检测对比例6得到的反应产物,检测结果为,对比例6得到的反应产物中5-甲基-N-丁基吡咯烷酮的产率为0%。
对比例7
按照实施例6的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是采用对比例1制备的以二氧化硅为载体的铂催化剂替换实施例6中实施例1制备的以Fe-S-1为载体的铂催化剂。
按照实施例8的检测方法检测对比例7得到的反应产物,检测结果为,对比例7得到的反应产物中5-甲基-N-丁基吡咯烷酮的产率为88%。
对比例8
按照实施例6的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是采用对比例2制备的以活性炭为载体的铂催化剂替换实施例6中实施例1制备的以Fe-S-1为载体的铂催化剂。
按照实施例8的检测方法检测对比例8得到的反应产物,检测结果为,对比例8得到的反应产物中5-甲基-N-丁基吡咯烷酮的产率为14%。
对比例9
按照实施例6的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是采用对比例3制备的以二氧化钛为载体的铂催化剂替换实施例6中实施例1制备的以Fe-S-1为载体的铂催化剂。
按照实施例8的检测方法检测对比例9得到的反应产物,检测结果为,对比例9得到的反应产物中5-甲基-N-丁基吡咯烷酮的产率为2%。
对比例10
按照实施例6的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是采用对比例4制备的以三氧化二铝为载体的铂催化剂替换实施例6中实施例1制备的以Fe-S-1为载体的铂催化剂。
按照实施例6的检测方法检测对比例10得到的反应产物,检测结果为,对比例10得到的反应产物中5-甲基-N-丁基吡咯烷酮的产率为41%。
对比例11
按照实施例6的技术方案制备得到5-甲基-N-丁基吡咯烷酮;不同的是采用对比例5制备的以HZSM-5分子筛为载体的铂催化剂替换实施例6中实施例1制备的以Fe-S-1为载体的铂催化剂。
按照实施例6的检测方法检测对比例11得到的反应产物,检测结果为,对比例11得到的反应产物中5-甲基-N-丁基吡咯烷酮的产率为4%。
以上实施例的说明只是用于帮助理解本发明的技术方案及其核心思想,应当指出,对于本技术领域的技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。