一种提高CRISPR/Cas9的同源重组修复效率的诱导剂、方法以及应用
文献发布时间:2023-06-19 19:28:50
技术领域
本发明属于基因工程技术领域,具体涉及一种提高CRISPR/Cas9的同源重组修复效率的诱导剂、方法以及应用。
背景技术
猪作为一种常见的大型实验动物,用于许多生物医学研究,特别是在转化研究领域。猪适合用于生物医学研究,因为(1)猪的的繁殖力高,很容易繁殖后代,(2)猪的生理结构、器官大小、基因组都与人具有极大的相似性,是一种非常适合用来模拟人生理特征和表型的模式动物,可以作为医学模型来研究疾病的发生与治疗,(3)猪具有经济价值,对于农业生产和遗传育种都有一定的价值。生产基因修饰猪模型可以快速的构建疾病模型猪和遗传育种优良的品质猪,这对于医学研究和农业发展都有重要的实际意义。
1985年,研究人员首次将DNA显微注射到受精卵中并产生出世界上第一头转基因猪,自此以后生产基因修饰猪的方法也随着基因工程技术的进步不断革新。猪的基因修饰技术通常涉及到外源基因的引入,包括使用逆转录病毒和慢病毒的策略,以及使用精子作为外源基因载体的方法。利用胚胎干细胞(ESCs)是生产转基因动物的理想策略,利用胚胎干细胞技术创造基因敲除小鼠的研究已被证实,然而在猪上还缺乏能够进行生殖系传递的ES细胞。体细胞核移植(SCNT)技术是猪基因工程学史上的重大突破,SCNT使用转基因(通过基因转移或基因敲除)核供体细胞来产生具有基因修饰的转基因个体。然而,通过同源重组对体细胞进行基因打靶操作复杂,效率极低。在基因组编辑技术被开发出来之前,基因敲除猪的生成都是相对罕见的。
随着基因编辑技术的快速发展,构建基因编辑猪模型的难度也大大降低。目前常用两种方法来生产基因编辑猪,细胞质注射和SCNT的方法。细胞质注射方法通常用于啮齿类动物,细胞质注射虽然操作简单,但是否成功获得了特定的基因型,只有在个体产生后才能显现出来。使用胞质注射的另一个实际问题是容易产生嵌合体个体,这种嵌合体个体混杂了突变细胞和无突变细胞,导致表型的不稳定表达。此外,嵌合体个体可以产生多种突变类型的生殖细胞。在下一代中选择具有所需基因型的个体需要大量的时间、劳动力和费用,这在生产妊娠期较长的大型动物时尤其成问题。相比之下,SCNT方法通过在核供体细胞中进行基因修饰,可以可靠地仅生产具有特定基因型的个体,大多数研究小组仍然采用SCNT生产基因修饰猪。
CRISPR/Cas9系统作为目前使用最为广泛的基因编辑技术,借助CRISPR基因编辑技术生产基因编辑猪模型的相关报道也越来越多。在农业上,借助基因编辑可以在短时间内培育出具有经济性状或具抗病性的农作物家畜品种,如重构猪UCP1基因产生的瘦肉型猪。2014年Whitworth,K.M.等利用CRISPR/Cas9成功将猪CD163(Cluster ofdifferentiation 163)基因敲除,CD163蛋白是猪蓝耳病病毒的受体,通过敲除CD163获得了免疫猪蓝耳病病毒感染的基因编辑猪模型。在医学研究上,2014年,Sato,M.等人对猪的GGTA1基因(α-1,3半乳糖苷转移酶)进行敲除,该基因敲除猪表现为超急性免疫排斥功能受损,能够有效降低异种器官移植过程中的免疫排斥反应,这对异种器官移植具有重要意义。2018年Yan等人利用CRISPR生产了HTT基因敲入的亨廷顿舞蹈症猪疾病模型。CRISPR技术在猪上的应用极大的加快了以猪模型为基础的的医学研究和农业育种进程。
CRISPR系统最初在原核生物中作为一种免疫外源核酸的防御机制被发现,经人工改造后,成为了广泛应用于各物种基因组编辑的强有力工具。CRISPR介导的基因编辑技术极大的推动了生物学基础研究,医学研究及农业育种的发展。然而CRISPR/Cas9系统的编辑能力仍在许多方面存在缺陷,例如CRISPR/Cas9系统介导的精确基因编辑效率较低,这主要是由于精确基因编辑依赖的同源重组修复(HDR)效率较低。
2018年Yan等人利用CRISPR生产了HTT(Huntingtin)基因敲入的亨廷顿舞蹈症猪疾病模型。精确敲入效率为9/2430(0.37%),张锋等人检测了CRISPR/Cas9系统在人类细胞中实现HDR介导的精确编辑效率约0.7%,多基因精确敲入的研究还没有相关报道,因为单个基因的敲入效率本身就低,研究者还在致力于解决如何提升单个基因的敲入效率,对于多基因敲入还没有相关尝试,但多基因编辑技术本身也是基因编辑中的一个重要课题。
发明内容
本发明的目的是为了提高CRISPR/Cas9的同源重组修复效率以及多基因编辑的效率的技术问题。
本发明提供一种提高CRISPR/Cas9的同源重组修复效率的诱导剂,所述诱导剂为M3814。
本发明提供上述的诱导剂在提高CRISPR/Cas9介导的编辑猪成纤维细胞的同源重组修复效率中的应用。
进一步地限定,所述M3814的有效浓度为0.1-2μM。
本发明一种提高CRISPR/Cas9介导的同源重组修复效率的方法,所述方法的步骤如下:将GenCrispr NLS-Cas9-NLS Nuclease,sgRNA和ssODN共转染到猪细胞中,添加M3814处理共转染后的猪细胞,培养至少72小时。
进一步地限定,所述M3814的浓度为0.1-2μM。
进一步地限定,所述猪细胞为猪成纤维细胞;所述ssODN为经过硫代修饰。
本发明提供上述的方法在编辑INS位点中的应用,对于1×10
本发明提供上述的方法在提高CRISPR/Cas9介导的编辑单基因或多基因的单位点或多位点的基因突变效率中的应用
进一步地限定,所述多基因为INS和RLN3;所述单基因为INS或RLN3;所述基因突变为碱基替换。
本发明提供上述的方法在提高CRISPR/Cas9介导的人源化修饰中的应用。
有益效果:本发明对CRISPR系统的精确基因编辑能力进行了优化,并将优化的CRISPR基因编辑技术应用在猪的成纤维细胞上。利用M3814可有效提高猪成纤维细胞中精准基因修饰的效率;M3814的最适施加浓度为2uM,可以提高精确编辑效率1.78倍,5uM及更高浓度的M3814会对猪成纤维细胞的存活率产生影响。利用硫代修饰的单链DNA模板可以使猪成纤维细胞中精准基因修饰的效率提升1.74倍。硫代修饰的单链DNA模板联合M3814进一步提升精准基因修饰的效率1.98倍。利用高效精准基因修饰方法可以实现在猪的成纤维细胞中对INS,RLN3基因进行高效的人源化修饰,并且可以实现同时对INS,RLN3两个基因进行人源化修饰。
在NHEJ介导的DNA修复中,DNA依赖的激酶(DNA-PKcs)会结合到断裂的DNA双链末端,保护断裂DNA末端免受切除,进而维持NHEJ修复通路对DSB进行修复。M3814是一种DNA依赖的激酶(DNA-PKcs)的抑制剂,本实验拟通过使用M3814作用于猪的成纤维细胞,抑制NHEJ通路,从而使DNA修复偏向于HDR修复,以此来提高HDR介导的精确基因编辑效率。在HDR介导的精确基因编辑中,需要同源修复模板的参与,在DSB处供体模板的浓度在一定程度上也影响着HDR介导的精确基因编辑,单链寡核苷酸形式的修复供体模板导入细胞后,在复杂的细胞环境中单链寡核苷酸模板极易被降解,对单链寡核苷酸模板进行硫代修饰能够极大的增强寡核苷酸模板的稳定性,从而避免因降解而带来的损失,使得更多的修复模板富集到DSB处,进而提高HDR的效率。
附图说明
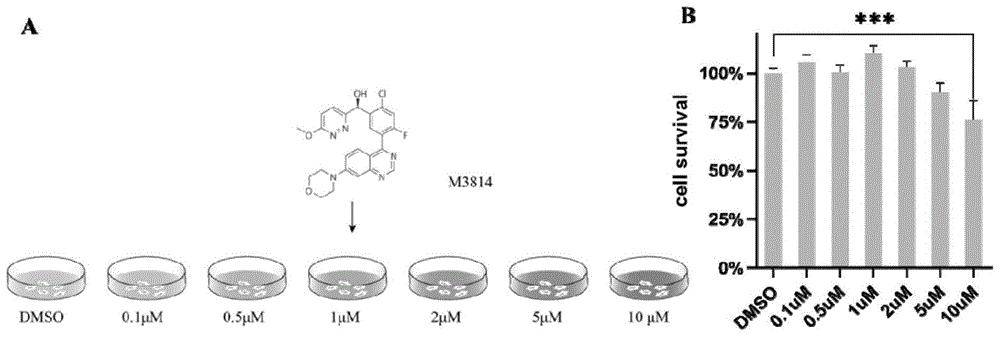
图1为不同浓度M3814对PEF细胞活性的影响;A:M3814的浓度对细胞毒性研究的实验流程。B:不同浓度的M3814对PEF细胞存活率的影响。
图2为RNP体外酶切鉴定sgRNA-INS的切割活性;A:PCR扩增靶DNA片段,对靶基因INS附近区域进行PCR扩增,片段大小为592bp;B:利用RNP体外酶切靶DNA片段(pINS 592)鉴定sgRNA-INS的活性,RNP对INS 592bp DNA片段进行消化产生了大小为263bp和329bp的两个片段。
图3为M3814浓度对HDR效率研究的流程。
图4为不同浓度M3814处理对HDR效率的影响;A:PCR扩增靶基因(INS)编辑区域得到建库测序的DNA片段(INS-255bp)B:不添加M3814处理处理时HDR编辑细胞占总突变细胞的比例;C,D,E,F,G:添加0.5μM,1μM,2μM,5μM,10μM M3814处理后HDR编辑细胞占总突变细胞的比例。H:不同浓度处理后HDR编辑细胞占总突变细胞的比例的倍数变化。
图5为硫代修饰的单链DNA模板对HDR效率研究的流程;A:硫代修饰单链DNA模板的示意图;B:探究硫代修饰的单链DNA模板对HDR效率研究的实验流程。
图6为不同单链DNA模板对HDR编辑的效率;A:利用普通模板后HDR编辑的效率;B:利用硫代修饰的单链DNA模板后HDR编辑的效率。
图7为硫代修饰的单链DNA模板联合M3814对HDR效率的影响;A:硫代修饰的单链DNA模板联合M3814后精确编辑的效率;B:硫代修饰的单链DNA模板联合M3814精确编辑效率的倍数变化。
图8为CRISPR介导的HDR精确编辑对猪INS基因进行人源化的设计;
图9为利用高效精确编辑方法对猪INS基因进行人源化的效率;A,B:检测猪INS基因精确编辑效率的高通量测序结果。A为使用优化前编辑方法进行精确编辑的效率;B为优化的精确编辑方法进行精确编辑的效率;C:使用优化前或优化的精确编辑方法后HDR精确编辑细胞占总突变细胞的比例。
图10为CRISPR介导的HDR精确编辑对猪RLN3基因进行人源化的设计;
图11为RNP体外酶切鉴定sgRNA-RLN3a及sgRNA-RLN3B的切割活性;A:RNP体外酶切鉴定sgRNA-RLN3a切割活性;B:RNP体外酶切鉴定sgRNA-RLN3b切割活性。
图12为利用高效精确编辑方法对猪RLN3基因进行人源化的效率;A,B:检测猪RLN3b位点精确编辑效率的高通量测序结果,A为使用优化前编辑方法进行精确编辑的效率,B为优化的精确编辑方法进行精确编辑的效率,C:使用优化前或优化的精确编辑方法后HDR精确编辑细胞占总突变细胞的比例。
图13为利用提高HDR效率策略对INS,RLN3两个位点同时人源化修饰的效率;A:对RLN3B和INS位点的编辑效率结果,左为RLN3B和INS位点单独编辑的效率,虚线框内为两个位点同时编辑的效率。B:对RLN3a和RLN3B位点的编辑效率结果,虚线框内为两个位点同时编辑的效率。C:对RLN3a和INS位点的编辑效率结果,虚线框内为两个位点同时编辑的效率。D:对RLN3a和RLN3B和INS位点的编辑效率结果,虚线框内为三个位点同时编辑的效率。
图14为利用M3814提高同源重组修复效率的技术路线;
图15为修饰模板+M3814处理提高同源重组效率的技术路线。
具体实施方式
Nedisertib为M3814别称。
表1
/>
(1)SOC液体培养基:称取TrypTone20 g,Yeast Extract 5g,NaCl 0.585g,KCl0.1864g,MgCl
(2)SOC固体培养基:向每100mLSOC液体培养基中添加1.5g琼脂粉,高压灭菌稍冷却后倒平板,待凝固后备用。
(3)TAE电泳液配制:Tris-Base 4.84g,冰乙酸1.142mL,0.5M EDTA(pH=8.0)0.4mL,加水补足1000mL。
(4)15%FBS的DMEM细胞培养液:FBS 7.5mL,NEAA 0.5mL,L-谷氨酰胺0.5mL,青霉素-链霉素0.5mL,丙酮酸钠0.5mL,加DMEM补足50mL。
表2引物序列
猪胎儿成纤维细胞的分离和培养:
(1)PEF细胞的复苏
取冻存有PEF细胞的冻存管在37℃水浴锅中快速震荡,将细胞从冻存管中吸取到装培养液的离心管中然后进行离心,1200rpm离心3min,去除上清后添加培养液,轻柔吹打重悬细胞沉淀,将细胞悬液接种到合适的培养皿中培养。
(2)PEF细胞的培养
用15%FBS的DMEM(1mM丙酮酸钠、2mM谷氨酰胺、0.1mM NEAA、1×双抗)培养液对PEF细胞进行培养。在37℃,5%CO
(3)PEF细胞的传代
细胞密度达到90%以上时需进行传代。弃掉旧的培养液加DPBS清洗,加37℃预热的0.25%EDTA胰酶消化2min,加两倍胰酶体积的培养液终止消化,用移液器轻轻吹打细胞后移入离心管,1200rpm离心3min,弃上清加培养液重悬细胞沉淀,按1:3的比例接种到适当培养皿中进行培养。
PEF细胞电转RNP:
(1)弃旧的PEF培养液,用DPBS清洗一遍,加0.25%EDTA胰蛋白酶消化2分钟,加两倍胰酶体积的培养液终止消化,用移液器轻轻吹打后转移到离心管,1200rpm离心3分钟,弃去上清液后加入培养液重新悬浮。对细胞进行计数,取1×10
(2)配置RNP混合液,对于1×10
(3)用RNP混合液重悬1×10
(4)电转后的细胞接种到合适的培养皿中继续培养,在培养细胞时加入M3814处理72h。
细胞活性检测:
使用Enhanced Cell Counting Kit-8检测细胞活性,主要操作步骤如下:
(1)将细胞接种到96孔板中,确保50%密度,24h后更换预混有M3814培养液,用含M3814的培养液处理细胞24h。
(2)每孔加入20微升增强型CCK-8溶液,在细胞培养箱内孵育0.5-4小时。
(4)使用酶标仪测定A450值。
CRISPR基因编辑结果鉴定:
对电转RNP以及加M3814处理后的细胞提取基因组DNA:参照基因组DNA提取试剂盒(TaKaRa)对PEF细胞基因组DNA进行提取,操作步骤如下
(1)收集PEF到1.5mL管中,加200μl无菌水重悬PEF细胞。
(2)加180μl Buffer GB、20μl的Proteinase K和10μl的RNase A吸打混匀,在56℃水浴中温浴裂解细胞。
(3)向裂解液中加200μl 100%乙醇。
(4)将吸附柱安置在收集柱上,将裂解溶液移至吸附柱中,12000rpm离心2min,弃滤液。
(5)加500μl WA Buffer至吸附柱中,12000rpm离心1min,弃滤液。
(6)加700μl WB Buffer至吸附柱中,12000rpm离心1min,弃滤液。
(7)重复操作步骤(6)。
(8)在12000rpm条件下空管离心2分钟。
(9)在吸附柱膜中央处加入50μL的灭菌水,静置5分钟。12000rpm离心2分钟洗脱DNA。
靶位点附近区域的DNA片段扩增:
(1)PCR扩增靶位点区
PCR反应体系如下
PCR反应程序如下
(2)DNA胶回收纯化
参照胶回收试剂盒(TaKaRa)进行DNA胶回收纯化
1)切碎胶块并称量胶重,计算转换体积。
2)加入溶解液GM Buffer溶胶。
3)在室温下使胶块充分溶解,可间断震荡加快溶解。
4)将溶解的胶溶液转移至吸附柱中,12000rpm离心1分钟,弃滤液,可分多次重复操作将胶溶液全部经过吸附柱Spin Column离心。
5)将700ul的WB Buffer加入吸附柱中,12000rpm离心30秒,弃滤液。
6)重复WB Buffer清洗操作步骤。
7)室温12000rpm离心1分钟,除尽残留液体。
8)将吸附柱放在新的1.5ml的离心管上,在吸附柱膜中央加30μl灭菌水,室温静置1分钟后12000rpm离心1分钟洗脱DNA。
Deep Seq测序及数据分析编辑效率:
将短片段胶回收DNA(200-300bp)送至浙江安诺优达公司进行建库测序。
测序数据的分析利用CRISPResso2完成。在Linux系统中运行CRISPResso2(Python2.7环境),运行命令行,调取测序文件(--fastq_r2),输入待比对基因的WT序列(--amplicon_seq),输入sgRNA序列信息(--guide_seq)输入HDR修复后的预期序列信息(-e)。利用CRISPResso2对高通量测序数据进行处理,得到靶位点基因的基因编辑结果的频率分析。
RNP体外酶切验证sgRNA活性:
(1)PCR反应制备目的基因的DNA片段。
(2)配制RNP体外酶切反应的体系如下,
将该体系放置室温孵育10min,使sgRNA与Cas9蛋白形成RNP复合体。将目的DNA稀释至50ng/μL,加入4μL目的DNA片段(200ng),将反应体系置于37℃培养箱进行酶切消化反应2h。
(3)将RNP酶切消化产物在2%的琼脂糖凝胶上进行电泳分析。
实施例1.一种提高CRISPR/Cas9介导的同源重组修复效率的方法
1.利用M3814提高同源重组修复效率的技术路线如图14所示,本实验对不同组别猪成纤维细胞的培养液中添加了不同剂量的M3814,包括0.1μM,0.5μM,1μM,2μM,5μM,10μM六组,并且以加入1‰DMSO作为对照。
使用含M3814的培养液培养猪成纤维细胞1天后,参照Cell Counting Kit-8试剂盒使用说明,利用酶标仪检测不同组别细胞的A450值,对细胞进行细胞活性分析(图1A)。结果显示,以对照组A450平均值设为1,0-2μM浓度的M3814不影响细胞的活性,施加5μM M3814使得细胞活性下降至90%,10μMM3814使得细胞活性下降至75%(图1B),表明5μM及更高浓度的M3814能对细胞产生一定的毒性。
M3814的浓度对HDR效率的影响:本次实验以猪的INS(Gene ID:397415)基因为靶位点,查询NCBI基因组数据库获取猪INS基因序列,参照sgRNA设计网站(
step1:将NCBI获取的序列粘贴至crispor网站genomic sequence窗口内,
step2:物种选择sus scrofa-Pig,
step3:PAM选择20bpNGG-spCas9,提交至网站,在网站输出结果中选择评分较高,且GC含量40-60%的sgRNA作为进行实验的sgRNA。设计并且交由公司化学合成了sgRNA-INS(表3所示),利用CRISPR RNP体外酶切反应验证sgRNA-INS的有效性。
2.RNP体外酶切验证sgRNA活性:
提取猪PEF细胞基因组DNA,使用引物INS592F,INS592R(表2所示)扩增靶位点附近区域,片段长度为592bp(>NC_010444.4:1497208-1497799Sus scrofa isolate TJTabasco breed Duroc chromosome 2,Sscrofa11.1,whole genome shotgun sequence,SEQ ID NO.1:
GGACACCACGGCCATGTCACTCGGAGACCAGCGGCACCCGGGCCTTGACTCCGTAAGATTCCTCCCTGGAGCCGCCTCGGGAGGAGGGGAGGGAAGGGGGGCTTCTCGAGCGGGACCGGGGCGTGGGAGGGGTGTCTCTGGGCTCAGCTCCCAGGGAGTTGGTCACTTTTACCAATTTCCTCCTTAAAAACTCCCCCGACCGCTCCCGGGACGCCCCCTCGGCTCACCCTGAGGGTTCTCCGCCTCCCGACGGGCCTTGGGCGTGTAGAAGAAGCCGCGCTCCCCGCACACCAGGTACAGCGCCTCCACCAGGTGGGAGCCGCACAGGTGCTGGTTCACGAAGGCCTGGGCCGGGGCGGGCGCCCAGAGGGCCAGCAGGGCCAGCAGGGGCAGGAGGCGCGTCCACAGGGCCATGGCGGGGGGTGAGGACCTGGGGGACGGGCGGCGTTGAGGCCAGCCAGGGGCACCTGCAGCCCCCACCAGCCTCACCCCCCAAGGGCTCCGAGAGAGGAGCCCACGTCCTCCTGCCGAGATGCCCTGGCCCCAGCAGGACAGCCCAGACCCAGCTGACGGGAGCCCAGGGGGACAGACC)(图2A),胶回收该DNA片段并经测序验证无误,得到了包含靶位点的DNA片段,将化学合成的sgRNA-INS与Cas9蛋白(GenCrispr NLS-Cas9-NLS Nuclease,Cat.No.Z03469)加入200μl微管中室温孵育10min,使其形成RNP复合体,之后向200μl管中加入包含靶位点的DNA片段,37°培养箱孵育2h后对反应产物进行凝胶电泳分析,结果显示,Cas9与sgRNA-INS复合体可以对包含INS靶位点的DNA片段片段产生切割(图2B),而不加入Cas9的对照组没有产生切割DNA条带,该实验证明了本实验所用的sgRNA-INS的对靶DNA具有活性。
3.PEF细胞电转RNP:
以INS为靶基因(Gene ID:397415),设计合成了116bp的单链寡核苷酸ssODN-INSA54T(sgRNA通过碱基互补配对方式与基因组DNA结合,sgRNA序列中包含20bp与靶基因同源互补序列,该同源互补区为CRISPR编辑系统的靶区域,以该区域为中心分别向5’和3’方向扩展40-50bp获得约120bp的序列,将此120bp序列中的特定位点进行根据实验需求的碱基突变,即可获得包含目标突变的ssODN序列,例:本实验中将INS基因中编码54位丙氨酸(A)的碱基gcc突变为编码苏氨酸(T)的碱基acg,有目的性的使猪INS基因进行人源化修饰(INSA54T),因为人的INS基因54位编码苏氨酸)表3所示),作为HDR修复中的模板DNA,单链模板中包含了2个碱基替换,期望在HDR介导的精确基因编辑作用下,对猪INS基因进行碱基替换类型的基因编辑。培养猪的成纤维细胞以1×10
GGCTTCTCGAGCGGGACCGGGGCGTGGGAGGGGTGTCTCTGGGCTCAGCTCCCAGGGAGTTGGTCACTTTTACCAATTTCCTCCTTAAAAACTCCCCCGACCGCTCCCGGGACGCCCCCTCGGCTCACCCTGAGGGTTCTCCGCCTCCCGACGGGCCTTGGGCGTGTAGAAGAAGCCGCGCTCCCCGCACACCAGGTACAGCGCCTCCACCAGGTGGGAGCCGCACAGGTGCTGGTTCACGAAGGCCTGGGCCGG)(图4A),短DNA片段经建库深度测序后,测序数据使用CRISPResso2进行基因编辑结果的分析处理,得到了不同处理组HDR基因编辑的效率。
深度测序结果显示,加DMSO组中6.37%的细胞基因组产生了突变,突变的细胞中约22.19%产生了HDR介导的精准基因修饰(图4B),0.5μM M3814处理成纤维细胞时,在16.07%突变的细胞中,约31.68%产生了HDR介导的精准基因修饰(图4C)。2μM M3814处理成纤维细胞时,在11.11%突变的细胞中,约39.59%产生了HDR介导的精准基因修饰(图4E),表明添加M3814处理提高了精准基因编辑的占比,相比于添加DMSO处理组,添加1μM时HDR的效率提升了1.52倍,添加2μM时HDR的效率提升了1.78倍(4H)。该实验结果表明使用M3814作用于成纤维细胞能够有效提升HDR的效率,添加2μM时提升效果最明显,且2μMM3814不会对细胞产生毒性。
实施例2.一种提高CRISPR/Cas9介导的同源重组修复效率的方法
1.利用化学合成得到了附加硫代修饰的单链寡核苷酸ssODN-INSA54T-M(表3所示)作为HDR修复通路中的供体模板,在硫代修饰的模板中5’端与3’端各三个核苷酸中磷酸基团中的一个PO键进行PS修饰(图5A)。修饰序列命名为ssODN-INSA54T-M,未修饰序列为ssODN-INSA54T。
2.PEF细胞电转RNP:对于1×10
用RNP混合液重悬1×10
利用电穿孔将Cas9,sgRNA-INS,ssODN-INSA54T-M共转染到成纤维细胞中,电转Cas9,sgRNA-INS,ssODN-INSA54T作为对照组(图5B),编辑后继续培养细胞72h,
将每组细胞作为一个细胞池提取基因组DNA,使用引物INS-A1,INS-S1对(表2所示)靶位点区域进行PCR扩增得到255bp DNA片段,短DNA片段经建库深度测序后,测序数据使用CRISPResso2进行基因编辑结果的分析处理,得到了不同处理组HDR基因编辑的效率。
测序结果表明,使用硫代修饰的寡核苷酸模板HDR介导的精确基因编辑效率为5.34%,相对于对照组,精准基因编辑的效率提升了1.74倍(图6)。
实施例3.一种提高CRISPR/Cas9介导的同源重组修复效率的方法
一、修饰模板+M3814处理提高同源重组效率的技术路线如图15所示,电转Cas9,sgRNA-INS,ssODN-INSA54T-M到成纤维细胞后,添加2μM M3814处理电转后的细胞72h。
对于1×10
用RNP混合液重悬1×10
硫代修饰的单链DNA模板与M3814联合使用时HDR效率为1.35%,HDR编辑细胞数占总突变细胞的43.97%(图7A),相比于未进行优化的精确基因编辑方法,联合使用策略可以提高HDR效率为1.98倍(图7B)。
将每组细胞作为一个细胞池提取基因组DNA,使用引物信息INS-A1,INS-S1对(表2所示)靶位点区域进行PCR扩增得到255bp DNA片段,短DNA片段经建库深度测序后,测序数据使用CRISPResso2进行基因编辑结果的分析处理,得到了不同处理组HDR基因编辑的效率。
结果表明联合使用硫代修饰的单链DNA模板与M3814处理是一种提高HDR效率的有效策略,这种策略对于在猪成纤维细胞中进行高效精确基因编辑具有一定潜力。利用提高HDR效率策略对INS位点进行人源化修饰:本实验将这种方法应用于在猪的成纤维细胞上对基因进行精确的碱基替换从而产生人源化修饰,经氨基酸序列分析猪INS氨基酸序列与人源氨基酸序列相差一个氨基酸,通过设计合成ssODN-INSA54T,供体模板中包含A54T突变,利用HDR介导的精确基因编辑,将A54T突变引入靶位点处产生人源化修饰(图8)。测序结果表明,在INS位点产生人源化修饰的HDR效率为0.83%(图9A),该细胞占总突变细胞的22.19%,使用优化的精确基因编辑方法产生人源化修饰的HDR效率为1.35%(图9B),该细胞占总突变细胞的43.97%,相比于对照组,精准编辑编辑效率提升了1.98倍(图9C)。
二、利用提高HDR效率策略对RLN3位点进行人源化修饰:选取了猪RLN3(Gene ID:503836)基因为靶点,因为经氨基酸序列分析猪RLN3氨基酸序列与人源氨基酸序列相差3个氨基酸,本研究设计合成了sgRNA-RLN3a,ssODN-RLN3a-N25S-M(表3所示),供体模板中包含N25S突变,利用HDR介导的精确基因编辑,将N25S突变引入RLN3a靶位点处产生人源化修饰。
同时合成了sgRNA-RLN3b,ssODN-RLN3b-S29AK34R-M(表3所示),该供体模板中包含S29A,K34R突变,利用HDR介导的精确基因编辑,将S29A,K34R突变引入RLN3b靶位点处(图10)。
利用RNP体外酶切实验,sgRNA-RLN3a与Cas9将PCR扩增(rln3a 553F+rln3a 553R引物对)得到的DNA片段RLN3a 553bp
(agtgtggcctgagctgaatcccaagcactcactcttttcatctttttgcaggggatgtcttctcagacacagattccaacgcagacagcgagttggacgaggcaatggcctccagcgaatggctggccctgaccaagtcccctgagaccttctatggggttcaaccaggctggcagagaacccctggggctcttaggggcagtcgtgatgtcctggctggcctctccagcagctgctgcaagtgggggtgcagcaagagtgaaatcagcagcctctgctagacaggggaccaggtggccatggagaaccagagtgaatgtcccagctctgctgtccacctaatgggtgtccaggtgggcacctgtttctagcccctcatgcattcattcatcagcaagtcacagagctcaggcactatgggctcagaatagagtcctcccacccaaccctgacctttggccagcctatcctgaccctgagcaggctgtgcccctacctggccacatggggaccctttctccccagcccatctcatgcctttcctggcctgtac,SEQ ID NO.3)切割为243bp与310bp两个片段(图11A),sgRNA-RLN3b与Cas9将PCR(RLN3B 607F+RLN3B 607R引物对)扩增得到的DNA片段RLN3b607bp(cgcttctgaacggcacataccagggttttgcaaacagcagccttgcctaatgcagggctcacacatagcaggtgttccagaaagagtagcctgttctctgacttttagctagggtgaatggcttgctgtccccacacacacaccaccccatcccagctcctctgctctgctaattggctcctccagccttggcacagctttgagggcttggctgtctccccagttgcatgtggagggaagggactctcttgctcttcatctcacctcccttcctggctcctcggacccttgtcctgtctcgtccaccccccttccccacacacattacctccccagcctcacccagagcttcatgggccagcatgtccgaccgtctccaccgggagcccccgcaggtaaagatgaccgctcggatgaattcacggccgcaaagcttcactccatagggtgacgcccgggcctcagtcctc agccacagctccccagccagcacccatacggccagcagcagcagcagcagtggacgtttggccattctggacgggctgcctgagatgtgagagtggacgcagcaaggcagctttctgatggttggtgcctcctggac,SEQ ID NO.4)切割为444bp与163bp两个片段(图11B),证明了sgRNA的活性,可以进行下一步的细胞水平基因编辑。
三、首先对RLN3a和RLN3b两个位点分别编辑:
对于1×10
用RNP混合液重悬1×10
将每组细胞作为一个细胞池提取基因组DNA,使用引物rln3a 258F,rln3a 258R对(表2所示)靶位点区域进行PCR扩增得到258bpDNA片段(gcaatggcctccagcgaatggctggccctgaccaagtcccctgagaccttctatggggttcaaccaggctggcagagaacccctggggctcttaggggcagtcgtgatgtcctggctggcctctccagcagctgctgcaagtgggggtgcagcaagagtgaaatcagcagcctctgctagacaggggaccaggtggccatggagaaccagagtgaatgtcccagctctgctgtccacctaatgggtgtccaggtgggc,SEQ ID NO.5)(图4A),短DNA片段经建库深度测序后,测序数据使用CRISPResso2进行基因编辑结果的分析处理,得到了不同处理组HDR基因编辑的效率。
四、对RLN3b位点编辑:
对于1×10
用RNP混合液重悬1×10
将每组细胞作为一个细胞池提取基因组DNA,使用引物RLN3B 272F RLN3B 272R引物对对(表2所示)靶位点区域进行PCR扩增得到272bpDNA片段(caaccatcagaaagctgccttgctgcgtccactctcacatctcaggcagcccgtccagaatggccaaacgtccactgctgctgctgctgctggccgtatgggtgctggctggggagctgtggctgaggactgaggcccgggcggcgccctatggagtgaggctttgcggccgtgaattcatccgagcggtcatctttacctgcgggggctcccggtggagacggtcggacatgctggcccatgaagctctgggtgaggctggggaggtaatg,SEQ ID NO.6)(图4A),短DNA片段经建库深度测序后,测序数据使用CRISPResso2进行基因编辑结果的分析处理,得到了不同处理组HDR基因编辑的效率。
通过电转RNP和ssODN-RLN3a-N25S-M(RNP和ssODN电转到PEF细胞中)对成纤维细胞进行RLN3a基因编辑。测序结果表明,对照组在RLN3a位点产生HDR编辑效率为0.04%,而使用优化方法后精确编辑效率为0.63%,通过电转RNP和ssODN-RLN3b-S29AK34R-M(表3所示),对成纤维细胞进行RLN3b基因编辑,在RLN3b位点产生人源化修饰的效率为1.64%,占所有突变细胞的18.86%,使用优化的精确基因编辑方法产生人源化修饰的效率为1.41%,占所有突变细胞的29.79%,相比于对照组,精准编辑编辑效率提升了1.58倍(图12)。
实施例4.单基因多位点编辑的方法
利用提高HDR效率策略对INS,RLN3两个位点同时人源化修饰:为了探究这种高效精准编辑方法的多基因编辑能力,分别进行了两个位点同时编辑,三个位点同时编辑。
一、两个位点同时编辑
1.通过电转sgRNA-INS,ssODN-INSA54T-M,sgRNA-RLN3a,ssODN-RLN3a-N25S-M,Cas9蛋白对INS和RLN3a两个位点进行编辑:100pmol sgRNA-INS+200pmol ssODN-INSA54T-M,100pmolsgRNA-RLN3a+200pmol ssODN-RLN3a-N25S-M,20μg Cas9蛋白对INS和RLN3a两个位点进行编辑,在室温下预混10min,最后用optiMEM补至20μL。
用RNP混合液重悬1×10
将每组细胞作为一个细胞池提取基因组DNA,使用引物INS-A1,INS-S1,RLN3a-258F,RLN3a-258R对(表2所示)靶位点区域进行PCR扩增得到INS255bp,RLN3a258bp片段(INS255bp,RLN3a258bp两个DNA片段单独测序,不混合),短DNA片段经建库深度测序后,测序数据使用CRISPResso2进行基因编辑结果的分析处理,得到了不同处理组HDR基因编辑的效率。
2.通过电转sgRNA-INS,ssODN-INSA54T-M,sgRNA-RLN3b,ssODN-RLN3b-S29AK34R-M,Cas9蛋白对INS和RLN3b两个位点进行编辑:100pmol sgRNA-RLN3a+200pmol ssODN-RLN3a-N25S-M,100pmol sgRNA-RLN3b+200pmol ssODN-RLN3b-S29AK34R-M,20μg Cas9蛋白对INS和RLN3b两个位点进行编辑,在室温下预混10min,最后用optiMEM补至20μL。
用RNP混合液重悬1×10
将每组细胞作为一个细胞池提取基因组DNA,使用引物INS-A1,INS-S1,RLN3b-272F,RLN3b-272R(表2所示)靶位点区域进行PCR扩增得到INS255bp,RLN3b272bpNA片段,短DNA片段经建库深度测序后,测序数据使用CRISPResso2进行基因编辑结果的分析处理,得到了不同处理组HDR基因编辑的效率。
3.通过电转sgRNA-RLN3a,ssODN-RLN3a-N25S-M,sgRNA-RLN3b,ssODN-RLN3b-S29AK34R-M,Cas9蛋白对RLN3a和RLN3b两个位点进行编辑:100pmol sgRNA-INS+200pmolssODN-INSA54T-M,100pmol sgRNA-RLN3b+200pmol ssODN-RLN3b-S29AK34R-M,20μg Cas9蛋白对RLN3a和RLN3b两个位点进行编辑,在室温下预混10min,最后用optiMEM补至20μL。
用RNP混合液重悬1×10
将每组细胞作为一个细胞池提取基因组DNA,使用引物RLN3b-272F,RLN3b-272R,RLN3a-258F,RLN3a-258R(表2所示)靶位点区域进行PCR扩增得到RLN3a-258bp,RLN3b-272bp,,短DNA片段经建库深度测序后,测序数据使用CRISPResso2进行基因编辑结果的分析处理,得到了不同处理组HDR基因编辑的效率。
二、三个位点同时编辑
通过电转sgRNA-INS,ssODN-INSA54T-M,sgRNA-RLN3a,ssODN-RLN3a-N25S-M,sgRNA-RLN3b,ssODN-RLN3b-S29AK34R-M,Cas9蛋白对INS和RLN3a和RLN3b三个位点进行编辑:100pmol sgRNA-INS+200pmol ssODN-INSA54T-M,100pmol sgRNA-RLN3a+200pmolssODN-RLN3a-N25S-M,100pmol sgRNA-RLN3b+200pmol ssODN-RLN3b-S29AK34R-M,30μgCas9蛋白对INS和RLN3a和RLN3b三个位点进行编辑,在室温下预混10min,最后用optiMEM补至20μL。
用RNP混合液重悬1×10
将每组细胞作为一个细胞池提取基因组DNA,使用引物INS-A1,INS-S1,RLN3b-272F,RLN3b-272R,RLN3a-258F,RLN3a-258R(表2所示)靶位点区域进行PCR扩增得到xxINS255bp,RLN3b-272bp,RLN3a-258bp,短DNA片段经建库深度测序后,测序数据使用CRISPResso2进行基因编辑结果的分析处理,得到了不同处理组HDR基因编辑的效率。
每组编辑的细胞作为一个细胞池检测多个基因的编辑效率,深度测序检测基因突变的分布模式,结果显示,同时对INS,RLN3a位点精确编辑的效率分别为1.99%,0.01%,同时对INS,RLN3b位点精确编辑的效率分别为3.0%,3.21%,同时对RLN3a,RLN3b位点精确编辑的效率分别为0.15%,0.68%,相比于单个位点编辑的效率,两个位点同时编辑的效率与单基因编辑没有明显差异。而进行同时对INS,RLN3a,RLN3b三个位点精确编辑的效率分别为0.29%,0.01%,0.04%,相比于单个基因精确编辑的效率,进行三个位点同时编辑时,HDR介导的精准基因修饰效率明显下降(图13)。
为了达到无痕编辑基因组的目的,没有采用质粒载体的形式,而是采用Cas9蛋白与sgRNA组合形成RNP对基因组进行编辑,采用RNP形式进行基因编辑具有响应时间短,脱靶效率低的优点,因为RNP进入细胞后会随着时间延长而逐渐降解失效,所以能够避免质粒载体整合入基因组破坏基因组的问题,真正实现无痕的精确基因编辑。由于本实验中采用了无载体的方式进行基因编辑,也就导致对于转染RNP的细胞没有进行富集筛选,为了消除转染的差异对基因编辑效率的影响,本实验通过测序数据的分析,单独研究了基因组突变细胞群中HDR基因编辑结果的占比。多个RNP与相应的ssODN共转染介导的多基因精确编辑的效率仍然较低,且同时对三个位点的精确基因编辑效率极低,可能是由于转染RNP的量过多对细胞的毒性作用引起的。
表3基因序列
/>
*Modification:2’-O-methyl and phosphorothioate modifications at thefirst three 5’and 3’terminal residue;ssODN-RLN3b-S29AK34R-M的对照组,加修饰用M后缀标注,不加修饰为ssODN-RLN3b-S29AK34R;ssODN-RLN3a-N25S-M的对照组,加修饰用M后缀标注,不加修饰为ssODN-RLN3a-N25S。
- 一种Cas9蛋白、CRISPR/Cas9系统、蘑菇基因编辑的方法及应用
- 一种可提高CRISPR/Cas9系统同源重组效率的方法和应用
- 一种提高CRISPR-Cas9基因编辑中同源重组修复效率的方法