一种普卡那肽的合成方法
文献发布时间:2023-06-19 19:30:30
技术领域
本发明涉及药物合成技术领域,具体涉及一种普卡那肽的合成方法。
背景技术
普卡那肽(Plecanatide,商品名为Trulance)为尿鸟苷蛋白(uroguanylin)的类似物,具有促尿钠排泄的鸟苷酸环化酶C(GC-C)受体激动剂的作用,可诱导液体转运到胃肠道,从而增加胃肠蠕动,治疗便秘。它由16个L型氨基酸残基组成,其中Cys4和Cys12、Cys7和Cys15分别通过二硫键相连接,其肽序列如下所示:
CN 108003222 A公开了一种普卡那肽的合成方法,通过固相和液相结合的方法来构建肽骨架,其中六肽在液相合成,并通过酶法将其偶联到肽树脂,完成肽链合成。在树脂上环化过程中,用1-5%的TFA在树脂上去除半胱氨酸保护基Mmt,然后在过氧化氢的帮助下环化,形成第一个二硫键。随后,通过在树脂上去除半胱氨酸巯基保护基团Trt,然后在DMF中碘的帮助下进行环化,形成第二个半胱氨酸二硫键。然而,六肽的液相合成以及它与树脂的偶联在商业上并不可行。
WO 2019215753 A2公开了一种组合使用固相和液相方法来合成普卡那肽的方法。该方法使用保护氨基酸来构建线性多肽骨架。通过利用CTC树脂在固相中合成保护的六肽和八肽片段,并将这些片段在液相中偶联,得到线性的多肽树脂。CN 109354607 A描述了普卡那肽的合成方法是先通过使用CTC树脂合成1-8位八肽片段,并且把该保护肽片段和八肽树脂偶联,随后裂解和两次定向环化得到普卡那肽粗品。然而,随着多肽片段长度的增加,溶解度将降低,偶联效率也将降低。因此,在液相和固相中偶联六肽和八肽在商业上是不可行的,并且会导致生成更多的杂质。经历这个反应过程,意味着很难获得99%以上的高纯度。
CN 107383170 A描述了使用固相法构建线性多肽骨架,将Trt基团用作所有四个半胱氨酸的巯基保护基,并通过随机氧化法一步形成两个二硫键的过程来合成普卡那肽。然而,通过这种方法容易形成更多错配的二硫键,并且会产生大量杂质,从而使纯化变得繁琐。与随机氧化法相比,分步氧化法在形成正确配对的二硫键方面更适合用于普卡那肽的氧化。
在现有报道的在分步氧化法中,专利文献CN104628827B、CN107383171A、WO2020034286A1公开StBu、Mmt和Acm基团被用作半胱氨酸的巯基保护基,在树脂上先选择性地脱去StBu/Mmt保护基,然后环合,接着在树脂上脱去Acm保护基后再次环合。然而,在树脂上一步脱保护Mmt保护基,需要重复使用TFA,它可能会部分切割树脂和不耐酸保护基团,如Trt、tBu和树脂,从而导致生成更多杂质,这些杂质反过来又会使纯化变得冗长;叔丁巯基(StBu)虽然可以用温和的还原剂去除,但由于脱保护时间过长,比如4-12小时,StBu不能用于常规固相多肽合成。在某些情况下,Acm基团被用作巯基保护基,但是有助于树脂上Acm基团去除和环化的条件反而会阻碍反应。因此,在树脂上进行两次氧化是不合适的。如果在液相中进行两次氧化,第一次反应的残余物、试剂和副产物都将继续进入下一阶段的反应中,增加产物纯化难度。
WO 2018205401 A1公开了一种通过保护氨基酸构建线性多肽骨架来合成普卡那肽的方法。该固相合成方法采用StBu基团作为在4和12位的半胱氨酸的巯基保护基,而在7和15位采用了3-氯丙氨酸。这个过程需要长时间的加热来去除StBu保护基,N-氯代丁二酰亚胺有助于树脂上二硫键的形成,第二个二硫键的形成是通过使用硫化剂在7和15位成环来完成的。然而,通过加热去除树脂上半胱氨酸的StBu保护基会导致一些副反应。此外,把3-氯丙氨酸转化为半胱氨酸二硫键的过程可能不完全,并且会导致生成一些杂质。因此,这一过程在商业上不可行。
因此,开发一种具有成本效益且商业上可行的普卡那肽合成方法是本领域技术人员需要解决的问题。
发明内容
本发明的目的在于提供一种可满足商业化规模生产的普卡那肽合成方法,该方法采用两步环合方法,第一步环合在固相上,第二步环合在液相里,以减少错配二硫键的形成以及降低产物纯化难度。在固相合成中插入合适的正交保护基,减少多肽杂质的生成,提高产物纯度。
为实现上述目的,本发明采用如下技术方案:
一种普卡那肽的合成方法,包括以下步骤:
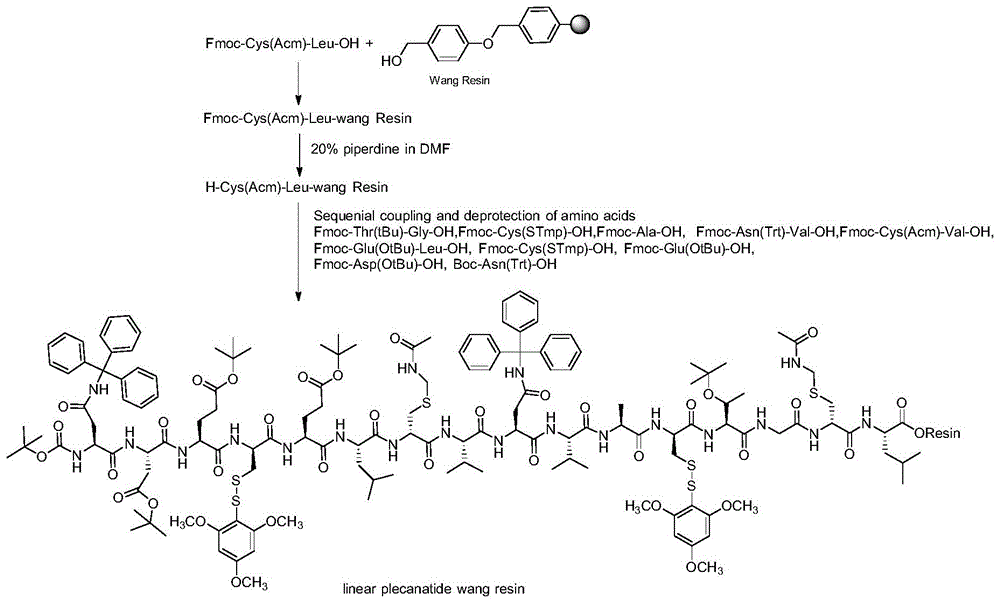
(1)按照SEQ ID NO:1所示氨基酸序列从C端到N端顺序,依次将N端和侧链偶联有保护基的氨基酸连接到树脂固相载体上,固相合成普卡那肽树脂,其中第4位和第12位半胱氨酸的侧链保护基为三甲氧基苯硫基,第7位和第15位半胱氨酸的侧链保护基为乙酰氨甲基;
(2)采用还原剂硫醇对普卡那肽树脂上的三甲氧基苯硫基进行脱保护,再利用N-氯代丁二酰亚胺进行环化形成第4、12位Cys的二硫键;
(3)利用酸裂解试剂切除普卡那肽树脂的固相载体以及肽链上除第7、15位之外的保护基,得到线性普卡那肽;
(4)采用含碘的乙酸对线性普卡那肽上的乙酰氨甲基进行脱保护,同时环化形成第7、15位Cys的二硫键,得到普卡那肽粗品;
(5)纯化,制得所述普卡那肽。
本发明对形成二硫键的两对半胱氨酸(Cys)使用两种不同的保护基,分步进行脱保护和环化。第一步的脱保护和环化在固相树脂上进行,方便该步骤产物单二硫键普卡那肽的纯化,本发明选用了三甲氧基苯硫基(S-Tmp)作为保护基,该保护基可以被温和的裂解试剂去除,而不会改变其他保护基。第二步的脱保护和环化在液相中完成,采用乙酰氨甲基(Acm)作为保护基。
步骤(1),利用固相合成法制备树脂肽。
所述固相合成方法包括:将Fmoc-Cys(Acm)-Leu-OH的羧基锚定到固相载体上,裂解脱去Fmoc基团,然后按照SEQ ID NO:1所示氨基酸序列顺序,依次将Fmoc-Thr(tBu)-Gly-OH、Fmoc-Cys(STmp)-OH、Fmoc-Ala-OH、Fmoc-Asn(Trt)-Val-OH、Fmoc-Cys(Acm)-Val-OH、Fmoc-Glu(OtBu)-Leu-OH、Fmoc-Cys(STmp)-OH、Fmoc-Glu(OtBu)-OH、Fmoc-Asp(OtBu)-OH、Boc-Asn(Trt)-OH进行偶联,得到Boc-Asn(Trt)-Asp(OtBu)-Glu(OtBu)-Cys(STmp)-Glu(OtBu)-Leu-Cys(Acm)-Val-Asn(Trt)-Val-Ala-Cys(STmp)-Thr(tBu)-Gly-Cys(Acm)-Leu-树脂。
为了降低合成成本和去除多肽杂质,本发明对普卡那肽的肽链结构进行有效分割,合理采用二肽片段即Fmoc-Cys(Acm)-Leu-OH、Fmoc-Thr(tBu)-Gly-OH、Fmoc-Asn(Trt)-Val-OH、Fmoc-Cys(Acm)-Val-OH、Fmoc-Glu(OtBu)-Leu-OH用于固相合成,该方式不仅简化工艺,提高多肽主链合成效率,而且有利于去除多肽杂质,提升产物纯度。
对于普卡那肽的合成,其Cys侧链保护基的选择关系着产物的纯度,本发明采用三甲氧基苯硫基(S-Tmp)作为第4位和第12位Cys侧链保护基,该保护基的去除条件较温和,可以选择性地脱去在固相上的巯基保护基,对其他保护基和树脂的影响小,在不改变其它保护基的情况下在树脂上形成二硫键。乙酰氨甲基(Acm)作为第7位和第15位Cys侧链保护基,研究表明,该保护基适合在液相条件下完成脱保护和环化反应。
本发明在合成第一位Asn时,采用Boc-Asn(Trt)-OH,其N端采用叔丁氧羰基(Boc)作为保护基,使得在后续酸裂解整体脱保护基更容易进行。
用于连接多肽羧基的固体载体品类繁多,如Wang树脂、CTC树脂、PHB树脂、HMPA树脂、HMPB树脂、Rink酸树脂、TentaGel TGA、TentaGel S PHB树脂。作为优选,所述树脂固相载体为Wang树脂或CTC树脂,取代度为1.0~1.2mmol/g,这两种树脂应用比较成熟且易得,并具有更好、更容易的锚固步骤。
在第一个氨基酸即Fmoc-Cys(Acm)-Leu-OH锚定后,使用溶于DMF、DMSO、N,N-二乙基乙酰胺、NMP或任何其他合适的非质子溶剂中的10-30%哌啶裂解Fmoc基团。
在切断Fmoc保护基后,游离胺基与序列中的下一个氨基酸偶联,即在DMF、DMSO、N,N-二乙基乙酰胺、NMP或任何其他合适的非质子溶剂中的Fmoc-Thr(tBu)-Gly-OH。
肽偶联试剂可以为DIC、HATU、HBTU、TBTU、PyBoP、HCTU的任意一种,再加上叔碱如DIEA或N-甲基吗啉进行联用。
各Fmoc氨基酸片段和偶联试剂总是过量加入,作为优选,其添加量均为肽树脂的2-4倍摩尔当量。
固相合成过程中通过Kaiser试验和四氯苯醌试验监测偶合和去偶合反应。
步骤(2)中,在固相树脂上进行第4、12位半胱氨酸巯基保护基S-Tmp的裂解及第一次环化。
作为优选,所述硫醇为二硫苏糖醇,与N-甲基吗啉联用裂解三甲氧基苯硫基。
作为优选,使用溶于DMF的含4.5%-6%二硫苏糖醇和0.08-0.12mol/L N-甲基吗啉对树脂上的三甲氧基苯硫基进行裂解。普卡那肽树脂与裂解液的混合比为200g:500mL。更为优选,裂解液中含5%的二硫苏糖醇和0.1mol/L N-甲基吗啉。
作为优选,裂解条件为室温下反应20-50分钟。裂解完成后,取固相,并用DMF洗涤多次。
往裂解后的肽树脂中加入溶于DMF的N-氯代丁二酰亚胺溶液进行环化。作为优选,N-氯代丁二酰亚胺的添加量为肽树脂的2-3倍摩尔当量。室温下反应20-50min。反应结束后,取固相,用DMF洗涤多次,再用甲醇收缩树脂。
步骤(3)中,利用酸裂解试剂切割多肽树脂以生成具有在4位和12位环化,7位和15位上带有未脱保护的Acm基团的线性普卡那肽骨架。
所述酸裂解试剂为三氟乙酸(TFA)、三异丙基硅烷(TIS)、乙二硫醇(EDT)和水以不同比例混合。作为优选,酸裂解试剂为三氟乙酸、三异丙基硅烷、乙二硫醇和水以体积比为92.5:2.5:2.5:2.5混合所得。
裂解条件为0~40℃下反应2~5小时。作为优选,10~20℃下反应3~4小时。
裂解反应完成后,收集滤液,加入到甲苯和异丙醚的混合液中,搅拌60分钟,过滤收集沉淀,然后用异丙醚进行打浆洗涤并干燥,得到带有Acm基团保护的(7位和15位)和一个环化(4位和12位)的多肽。
步骤(4)中,在液相条件下,使用加碘的乙酸将巯基保护基Acm去除,同时完成第二次环化,从而得到普卡那肽的粗品。
作为优选,在15±2℃条件下将含碘的乙酸滴加到线性普卡那肽中,滴加完毕,升温至室温,再反应至起始物料的含量降至0.1%以下。通过超高效液相色谱(UPLC)分析监控反应完成情况。反应时间为5-6小时,然后用抗坏血酸终止反应。
作为优选,乙酸溶液中碘的浓度为0.18mol/L。碘的添加量为线性普卡那肽的1.5倍摩尔当量。
反应完成后,将被稀释的反应物料进行纳米过滤,以降低溶液体积,同时除去低分子量的杂质,得到普卡那肽粗品。
步骤(5)中,将普卡那肽粗品经过两个阶段的反相制备色谱纯化,最后冷冻干燥。
纯化分两步进行,第一步:将普卡那肽粗品通过C18反相柱,以0.1%的三氟乙酸水溶液为流动相A,乙腈为流动相B进行洗脱,收集目标峰馏分;第二步:将收集的目标峰馏分装入C18反相柱,用含质量百分比0.15%的醋酸铵水溶液洗涤,再以含体积比0.1%的醋酸水溶液为流动相A,乙腈为流动相B进行洗脱,收集目标产物。
本发明具备的有益效果:
(1)本发明通过固相合成法制备普卡那肽,合成过程中,通过对多肽链合理分割,利用五个二肽片段构建多肽骨架,简化合成工艺的同时减少多肽杂质生成。
(2)本发明选择STmp和Acm作为半胱氨酸正交的巯基保护基,其中4位和12位的半胱氨酸用STmp保护基修饰,7位和15位的半胱氨酸用Acm保护基修饰。分步环化过程中,第一步在固相树脂中脱去STmp保护基并形成第一个二硫键,该步骤中脱保护条件温和,避免对其他保护基和树脂的断裂;第二步在液相中脱去Acm保护基并形成第二个二硫键。上述两步环化方式反应效率高,产物杂质少。
(3)本发明提供的合成方法工艺操作简单,反应条件温和,产物经反相色谱纯化纯度达到99.5%,单个最大杂质含量小于0.1%,满足商业化规模生产的要求。
附图说明
图1为实施例1中Fmoc-Thr(tBu)-Gly-OH的合成反应示意图。
图2为实施例1中普卡那肽树脂的制备示意图。
图3为实施例1中普卡那肽树脂上第一个二硫键形成的反应示意图。
图4为实施例1中单二硫键的普卡那肽Wang树脂的裂解反应示意图。
图5为实施例1中普卡那肽粗品形成的反应示意图。
图6为对比例1中普卡那肽树脂的制备示意图。
图7为对比例1中普卡那肽Wang树脂的裂解反应示意图。
图8为对比例1中普卡那肽粗品形成的反应示意图。
具体实施方式
下面结合具体实施例对本发明做进一步说明。以下实施例仅用于说明本发明,不用来限制本发明的适用范围。在不背离本发明精神和本质的情况下,对本发明方法、步骤或条件所做的修改或替换,均属于本发明的范围。
下述实施例中所使用的试验方法如无特殊说明,均为常规方法;所使用的材料、试剂等,如无特殊说明,为可从商业途径得到的试剂和材料。
缩写说明:
Acm:乙酰氨甲基;
Boc:叔丁氧羰基;
CTC:2-氯三苯甲基氯;
DCC:N,N'-二环己基碳二亚胺;
DCM:二氯甲烷;
DIC:N,N-二异丙基碳二亚胺;
DIPEA:N,N-二异丙基乙胺;
DMAP:4-二甲氨基吡啶;
DMF:N,N-二甲基甲酰胺;
DMSO:二甲基亚砜;
DTT:二硫苏糖醇;
EDT:1,2-乙二硫醇;
Fmoc:9-芴基甲氧基羰基;
HATU:2-(7-偶氮苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯;
HBTU:苯并三氮唑-N,N,N',N'-四甲基脲六氟磷酸盐;
HCTU:6-氯苯并三氮唑-1,1,3,3-四甲基脲六氟磷酸酯;
HOBt:1-羟基苯并三唑;
IPA:异丙醇;
MTBE:甲基叔丁基醚;
MeOH:甲醇;
ΝMΡ:N-甲基-2-吡咯烷酮;
PyBOP:1H-苯并三唑-1-基氧三吡咯烷基六氟磷酸盐;
StBu:S-叔丁巯基;
STmp:三甲氧基苯硫基;
TBTU:O-苯并三氮唑-N,N,N',N'-四甲基脲四氟硼酸酯;
TFA:三氟乙酸;
TIS:三异丙基硅烷;
THF:四氢呋喃;
Trt:三苯甲基。
实施例1
一、二肽合成
Fmoc-Thr(tBu)-Gly-OH的合成
a)Fmoc-Thr(tBu)-OSu的合成
称取Fmoc-Thr(tBu)-OH(50.0g,125.8mmol)并加到含有400mL四氢呋喃的三颈烧瓶中。将此混合物在25±2℃下搅拌5分钟,然后加入17.37g(150.9mmol)N-羟基丁二酰亚胺,并搅拌5-10分钟。在一个单独的圆底烧瓶中加入31.14g(150.9mmol)的DCC,并加入100mL四氢呋喃搅拌溶解。然后在10±2℃的条件下将该DCC的THF溶液慢慢滴加至上述Fmoc-Thr(tBu)-OH溶液中,滴加时间控制在30-45分钟。滴加完成后,将系统加热至室温并再反应3小时。通过薄层色谱法(TLC)监控反应,直至原料完全反应。过滤反应液以除去尿素,并通过旋转蒸发仪完全除去THF以获得胶状的固体产物。向上述胶状固体产品中加入400mL异丙醚,并搅拌30分钟,然后过滤。过滤和真空干燥后,得到54.0g白色固体,产率为86.7%。
b)Fmoc-Thr(tBu)-Gly-OH的合成
称取L-甘氨酸(9.1g,121.3mmol)加入到装有300mL四氢呋喃和水(1:1)的三颈瓶中。将该混合物在25±2℃下搅拌5分钟,然后加入12.86g(121.3mmol)Na
上述反应进程如图1所示。
其余四种二肽,即Fmoc-Cys(Acm)-Leu-OH,Fmoc-Cys(Acm)-Val-OH,Fmoc-Asn(Trt)-Val-OH,Fmoc-Glu(OtBu)-Leu-OH,均按上述方案制备。
二、采用分步固相合成法制备肽树脂
1、取代度为0.9mmol/g的Fmoc-Cys(Acm)-Leu-Wang树脂的合成
称取50.0g取代度为1.1mmol/g的Wang树脂,并将其加到固相多肽合成仪中。随后,用DMF洗涤树脂两次,并在DMF中溶胀30分钟。然后将58.04g Fmoc-Cys(Acm)-Leu-OH、14.85g HOBt和14.2g DIC溶解在DMF中,并在冰水浴下搅拌5-10分钟。将此混合物加到上述装有树脂的合成仪中。5分钟后,加入0.1g DMAP并搅拌2小时。接着,用DMF和DCM分别洗涤树脂3次,并用醋酸酐/吡啶/DMF(15mL:15mL:500mL)封闭30分钟,然后排干树脂。用甲醇收缩树脂后,将甲醇抽干,以获得取代度为0.9mmol/g的Fmoc-Cys(Acm)-Leu-Wang树脂。
2、线性普卡那肽Wang树脂的制备
称取68.0g取代度为0.9mmol/g的Fmoc-Cys(Acm)-Leu-Wang树脂,并将其加到固相多肽合成仪中。随后,使用DMF洗涤Fmoc-Cys(Acm)-Leu-Wang树脂两次,并在DMF中溶胀30分钟。使用溶于DMF的20%哌啶去除Fmoc保护,然后使用DMF洗涤树脂5次。树脂通过茚三酮试验进行测试,其中Fmoc的去除通过树脂颜色的变化来判定。
将61.36g Fmoc-Thr(tBu)-Gly-OH(135.0mmol)、HOBt(18.22g,135.0mmol)溶于DMF中,并将此混合物加到固相反应合成仪中,再加入DIC(17.0g,135.0mmol)并在室温下反应3-4小时。通过茚三酮试验确定反应终点。
上述Fmoc去保护步骤和相应的氨基酸偶联步骤是基于普卡那肽的多肽骨架序列重复进行的,按照Fmoc-Cys(STmp)-OH、Fmoc-Ala-OH、Fmoc-Asn(Trt)-Val-OH、Fmoc-Cys(Acm)-Val-OH、Fmoc-Glu(OtBu)-Leu-OH、Fmoc-Cys(STmp)-OH、Fmoc-Glu(OtBu)-OH、Fmoc-Asp(OtBu)-OH、Boc-Asn(Trt)-OH的顺序依次偶合的。反应进程如图2所示。
在所有氨基酸完成偶联后,用甲醇收缩树脂,并将树脂排干,获得220g线性普卡那肽Wang树脂。
三、线性普卡那肽Wang树脂上S-Tmp保护基的裂解
称取200.0g在步骤2中获得的取代度为0.9mmol/g的线性普卡那肽Wang树脂,并将其加入到固相合成反应仪中。随后,使用DMF洗涤线性普卡那肽Wang树脂两次,并在DMF中溶胀30分钟。将5.5mL N-甲基吗啉、25g二硫代苏糖醇溶于500mL DMF中,将此混合液装入固相反应合成仪中并搅拌30分钟。反应完成后,将溶液排干,并用DMF洗涤四次。
四、通过在树脂上的环化得到第一个二硫键
向步骤三中获得的肽树脂中加入溶于DMF(500mL)的N-氯代丁二酰亚胺(12.0g)溶液,并在室温下反应30分钟。反应完成后,将溶液排干并用DMF洗涤六次,然后用甲醇收缩树脂。将树脂干燥后,得到190g单二硫键的普卡那肽Wang树脂。反应进程如图3所示。
五、单二硫键的普卡那肽Wang树脂的裂解
以TFA:EDT:TIS:H
六、普卡那肽粗品的合成
通过加碘的乙酸在液相中脱除巯基保护基Acm并环合二硫键
将碘(11.4g,90.35mmol)溶于乙酸(500mL)中,然后在15±2℃条件下慢慢滴加到步骤五中获得的含有单二硫键的普卡那肽(110g,60.23mmol)中,滴加时间控制在30-40分钟。滴加完成后,将系统升温至室温,再反应5-6小时。通过UPLC(超高效液相色谱)分析监控反应完成情况,直到起始物料(单二硫键多肽)的含量降至0.1%以下,然后反应用抗坏血酸溶液猝灭。反应进程如图5所示。
反应完成后,将被稀释的反应物料进行纳米过滤,以降低溶液体积,同时除去低分子量的杂质。
七、普卡那肽的纯化
第一阶段
将从步骤六获得的多肽粗品溶液装载到C18反相柱上,并用流动相进行洗脱,其中流动相A为0.1%TFA水溶液,流动相B为乙腈。收集纯度大于99%的组分作为目标馏分。
第二阶段
将从第1阶段获得的主馏分装载到C18反相柱上,用0.15%醋酸铵溶液洗涤60分钟,然后用流动相洗脱,其中流动相A为0.1%醋酸水溶液,流动相B为乙腈。将纯度大于99%且单个最大杂质<0.1%的组分收集起来,再经过冷冻干燥,以获得纯普卡那肽。
产量:45.0g,HPLC纯度:>99.5%。
HRMS(ESI)m/z:C
对比例1
本对比例采用三苯甲基(Trt)作为第4位和第12位Cys侧链保护基,第7位和第15位Cys侧链保护基保持不变,继续使用乙酰氨甲基(Acm)保护基。
一、分步固相合成法制备多肽树脂
合成方法参照实施例1,区别在于合成第4位和第12位Cys时采用Fmoc-Cys(Trt)-OH,反应进程如图6所示。
在所有氨基酸偶联后,用甲醇收缩树脂,并将其排干,获得线性普卡那肽Wang树脂220g。
二、线性普卡那肽的合成
1.1L裂解试剂按体积比TFA:EDT:TIS:H
三、普卡那肽粗品的合成
i)使用过氧化氢进行第一次环化:
向步骤二中获得的线性普卡那肽(105g)加入105L的水,并通过氨水溶液将溶液的pH值调节至8.0-8.3。然后在25±2℃的温度下,向上述溶液中逐滴加入90mL的30%过氧化氢溶液。控制反应在25±2℃的条件下搅拌60-120分钟,并通过UPLC监控反应进程。反应完成后,在不进一步分离固体的情况下直接进行下一步反应。
ii)在使用溶于乙酸的碘进行二次环化:
在一个单独的圆底烧瓶中,将12.5g碘溶于1500mL 50%乙酸水溶液中。在15±2℃的温度下,逐滴加入到上述第一次环化的产物溶液中,并使之在30-45分钟内滴加完成。滴加完成后,将系统升温至室温,并再反应5-6小时。通过UPLC监控反应进程,直至反应完成。反应完成后,用抗坏血酸溶液猝灭。然后,用NaHCO
四、普卡那肽的纯化
第1阶段:将从步骤三获得的多肽粗品溶液通过C18反相柱,并用流动相进行洗脱,其中流动相A为0.1%TFA水溶液,流动相B为乙腈。
第2阶段:将从第1阶段获得的主馏分装载在反相C18柱上,用0.15%醋酸铵溶液洗涤60分钟,然后用流动相洗脱,其中流动相A为0.1%醋酸水溶液,流动相B为乙腈。
将纯度大于99%且单个最大杂质<0.1%的组分收集起来,再经过冷冻干燥,以获得纯普卡那肽。产量:25.0g。
结论:本对比例中不使用STmp保护基,无法实现通过在树脂上先环化形成一个二硫桥,再液相环化法形成第二个二硫健。只能在液相中采用不同的条件完成两次环化,第一个环化反应的副产物和杂质将转入下一个环化阶段,大大降低了普卡那肽粗品的纯度。因此,纯化变得困难,产率急剧下降。
- 一种纳米金的绿色可控合成方法及其一步肽功能化方法
- 一种基于Fmoc二肽的合成阿拉瑞林的方法
- 一种普利卡那肽的固相片段合成方法
- 一种禽流感合成肽疫苗及其制备方法
- 一种索马鲁肽的合成方法
- 一种通过二次环化固相合成普卡那肽的方法
- 一种普卡那肽的合成方法