一种聚酰胺-聚酰亚胺复合物及其制备方法和应用
文献发布时间:2024-04-18 19:58:21
技术领域
本发明属于聚酰亚胺材料制备和应用领域,具体涉及一种聚酰胺-聚酰亚胺复合物及其制备方法和应用。
背景技术
柔性透明衬底是柔性透明电子器件的重要组成部分,作为柔性透明显示用的衬底需要具备以下几个条件:
(1)具有良好的耐热性;
(2)具有良好的耐有机溶剂性;
(3)具有良好的透光性。
透明聚酰亚胺(PI)由于具有高热稳定性、高透光率等优点,被认为是柔性显示领域最具潜力的衬底材料。同时,与无机玻璃相比,透明聚酰亚胺更兼具柔韧性、低密度和易加工性,使其备受柔性显示领域的青睐。为获得具有高玻璃化转变温度、低热膨胀系数、高透光性和良好力学性能的无色透明的聚酰亚胺,现有的主流制备方法包括以下几种:
1)纳米填料原位聚合法合成聚酰亚胺复合体系,该方法将TiO
2)无规共聚法合成聚酰亚胺-聚酰胺体系,该方法将含有氢键的芳香族聚酰胺通过无规共聚的方式与聚酰亚胺聚合制备出单链型聚酰亚胺-聚酰胺聚合物;通过该方法制备的聚酰亚胺-聚酰胺聚合物的氢键排列规整性较差,大大降低氢键的作用,导致制备的聚酰亚胺-聚酰胺薄膜整体性能较差,热膨胀系数较大、黄度和雾度都偏大;
3)嵌段共聚法合成聚酰亚胺-聚酰胺体系,该方法通过在溶解了二胺单体的前驱体聚酰胺酸溶液中引入氢键聚合物单体得到聚酰胺酸-聚酰胺聚合物,再对聚酰胺酸-聚酰胺聚合物进行亚胺化反应,制备出单链型的聚酰亚胺-聚酰胺聚合物;请参阅图1,通过该方法制备的聚酰亚胺-聚酰胺聚合物的氢键排列规整,但降低了聚酰亚胺的亚胺环密度且不能充分利用联苯结构和聚酰胺链间的氢键,进而削弱了聚酰亚胺与聚酰胺的相互作用,导致聚酰亚胺-聚酰胺体系的热性能不佳。
发明内容
本发明的目的在于提供一种基于原位聚合法向可溶性聚酰亚胺基体混入芳香聚酰胺增强相制备聚酰胺-聚酰亚胺复合材料的方法。
本发明所述的一种聚酰胺-聚酰亚胺复合物的制备方法,包括下述步骤:
将二胺单体溶解于可溶性聚酰亚胺溶液中,并向所述溶液缓慢加入至少一种引入氢键作用的聚合物单体,生成芳香聚酰胺分子链,所述芳香聚酰胺分子链通过原位聚合与聚酰亚胺分子链实现化学共混,得到芳香聚酰胺-聚酰亚胺双链共混聚合物。
与现有技术相比,本发明提供的基于原位聚合法向可溶性聚酰亚胺基体中混入芳香聚酰胺增强相制备聚酰胺-聚酰亚胺复合物的方法,可制备出具有新的结构的聚酰胺-聚酰亚胺复合物,该复合物含有双链结构且双链的链间具有互穿的氢键网络结构。同时,该制备方法简单,所用原材料、制备设备等均为本领域熟知的,经济效益高。
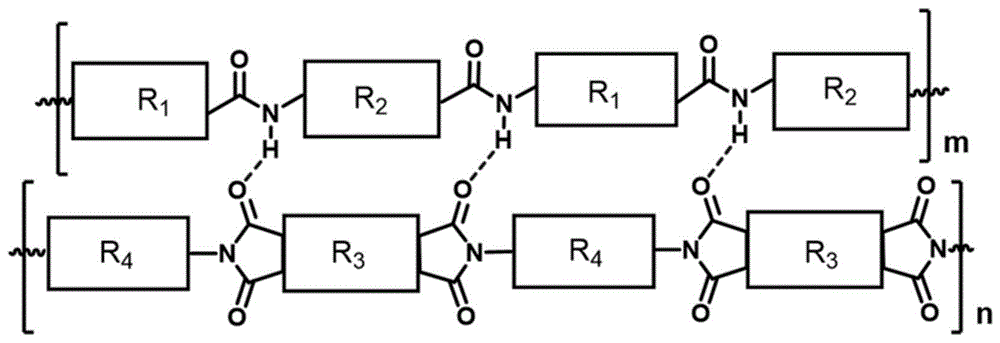
同时,根据本发明所述的制备方法制备出的一种聚酰胺-聚酰亚胺复合物,所述复合物具有由芳香聚酰胺分子链、聚酰亚胺分子链形成的双链结构,且所述双链的链间具有互穿的氢键网络结构,其结构式为:
其中,m和n为正整数,代表聚合度,5≤m≤100,10≤n≤550,R
相对于现有技术,本发明所述聚酰胺-聚酰亚胺复合物含有的双链结构,能充分利用有序氢键与亚胺环的作用且不破坏聚酰亚胺的结构,双链链间互穿的氢键网络结构极大的削弱芳香聚酰胺分子链与聚酰亚胺分子链的相互运动能力并减小双链间的链间距,使聚酰亚胺基体与芳香聚酰胺增强相之间的相互作用力进一步增强;同时,由于有序氢键与亚胺环作用能有效避免芳香聚酰胺分子间因有序氢键引起的聚合物链结晶,可使聚酰亚胺基体中形成分子水平分散的刚性聚酰胺聚合物,最大限度发挥刚性聚酰胺大分子对聚酰亚胺基体的增强效果。因此,含有双链且链间具有互穿的氢键网络结构的聚酰胺-聚酰亚胺复合物,具有更低的热膨胀系数、更高的玻璃化转变温度和更优异的尺寸热稳定性。
进一步地,所述聚酰亚胺为可溶性聚酰亚胺。
进一步地,所述芳香聚酰胺分子链中的R
以上各结构式中的R
以上各结构式中的R
其中,~波浪线表示连接键,P为正整数。
进一步地,所述芳香聚酰胺分子链的R
以上各结构式中的R
其中,虚线代表形成环状连接方式。
进一步地,所述聚酰亚胺分子链中的R
其中,虚线代表形成环状连接方式。
同时,本发明所述聚酰胺-聚酰亚胺复合物制备的薄膜,可适用于柔性电子器件的基底和盖板,如柔性碳纳米管晶体管器件、柔性氧化物半导体器件和柔性传感器等的基底;如柔性有机发光二极管(OLED)屏、柔性太阳能电池等的盖板。
与现有技术相比,基于本发明提供的聚酰胺-聚酰亚胺复合物制得的薄膜具有更低的热膨胀系数、更高的玻璃化转变温度,进而拥有优异的尺寸热稳定性,使薄膜在器件加工过程中不易卷曲或剥离,在高温气氛中加工时不发生尺寸变化甚至熔化。
为了更好地理解和实施,下面结合附图详细说明本发明。
附图说明
图1嵌段共聚法合成聚酰亚胺-聚酰胺的骨架结构式;
图2为本发明聚酰胺-聚酰亚胺复合物的骨架结构式;
图3为本发明聚酰胺-聚酰亚胺复合物的双链链间互穿网络结构示意图;
图4为本发明聚酰胺-聚酰亚胺复合物的合成过程图;
图5为本发明实施例1-4和对比例在50~400℃温度范围内升温速率为10℃·min
图6为本发明实施例1-4和对比例的聚酰胺在氮气氛下升温速率为10℃·min
图7为本发明实施例1-4和对比例在升温速率为10℃·min
图8为本发明实施例1-4和对比例在数据采集温度范围为50至250℃时,热膨胀系数下降百分比;
图9为本发明实施例1-4和对比例的光学透射光谱,插图是实施例5覆盖在图片上的薄膜光学示意图。
具体实施方式
为了便于理解本发明,下文将对本发明做更全面、细致地描述,但本发明的保护范围并不限于以下具体实施例。
除非另有定义,下文中所使用的所有专业术语与本领域技术人员通常理解含义相同。本文中所使用的专业术语只是为了描述具体实施例的目的,并不是旨在限制本发明的保护范围。
除非另有特别说明,本发明中用到的各种原材料、试剂、仪器和设备等均为本领域熟知的,但不限制本发明的实施,其他本领域熟知的一些试剂和设备都可适用于本发明以下实施方式的实施。
芳香族聚酰胺具有优异的热稳定性和力学性能,通常以聚合物的形式通过物理共混的方式引入聚酰亚胺体系中作为增强相,但通过物理共混的聚酰胺与聚酰亚胺之间不能形成较强的氢键相互作用,导致此类聚酰亚胺复合材料的界面相互作用较差,热性能的增强效果不理想。
因此,本发明重新研究了一种将性能优异的芳香族聚酰胺引入聚酰亚胺体系中作为增强相的混合方法。基于芳香族聚酰胺分子链与联苯结构之间存在较强的氢键相互作用,本发明研究利用原位聚合法,在溶解有二胺单体的可溶性聚酰亚胺基体溶液中加入引入氢键的聚合物单体原位进行增强相聚酰胺分子链的聚合,使聚合生成的芳香聚酰胺分子链与聚酰亚胺分子链实现化学共混,得到芳香聚酰胺-聚酰亚胺双链共混聚合物。该方法能充分利用有序氢键与亚胺环的作用且不破坏聚酰亚胺的结构,极大的削弱芳香聚酰胺链与聚酰亚胺链的相互运动能力并减小双链间的链间距,使聚酰亚胺基体与芳香聚酰胺增强相之间的相互作用力进一步增强;同时,由于有序氢键与亚胺环作用能有效避免芳香聚酰胺分子间因有序氢键引起的聚合物链结晶,可使聚酰亚胺基体中形成分子水平分散的刚性聚酰胺聚合物,最大限度发挥刚性聚酰胺大分子对聚酰亚胺基体的增强效果。
以下具体说明本申请具有双链共混的聚酰胺-聚酰亚胺复合物的制备方法,包括以下步骤。
(一)制备前驱体聚酰胺酸
在极性非质子溶剂中加入二胺单体与二酐单体,在0℃~25℃下的惰性气氛中连续搅拌反应生成包含R
所述的极性非质子溶剂选自:N-甲基-2-吡咯烷酮、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、γ-丁内酯等。
所述的R
所述的R
以上各结构式中的R
控制所述二胺单体与二酐单体的摩尔比在(1±0.02)。
控制所述二胺单体与二酐单体溶于极性非质子溶液中时,溶液的固含量在10%~20%。
控制所述二胺单体与二酐单体在极性非质子溶液中反应的时间在12h~24h。
(二)制备可溶性聚酰亚胺固体
在所述前驱体聚酰胺酸中加入酰化试剂,生成聚酰亚胺胶液,并将所述聚酰亚胺胶液置于乙醇中沉降,并用乙醇抽提沉降物,经真空干燥得到可溶性聚酰亚胺固体。
所述酰化剂选自:常用的酰化剂,如乙酸酐和吡啶。
控制所述酰化剂与聚酰胺酸的摩尔比,即mol
控制所述前驱体聚酰胺酸中加入酰化试剂进行酰化反应的时间在24h~48h。
(三)制备聚酰亚胺基体溶液
将所述可溶性聚酰亚胺固体溶于极性非质子溶液中,配置成含聚酰亚胺分子链的聚酰亚胺溶液,并在所述聚酰亚胺溶液中加入二胺单体,待二胺单体溶解完全后,得到聚酰亚胺基体溶液。
所述的极性非质子溶剂选自:N-甲基-2-吡咯烷酮、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、γ-丁内酯等。
控制所述聚酰亚胺溶液的固含量在5%~30%。
需要说明的是,本申请步骤(一)、步骤(二)提供一种可溶性聚酰亚胺固体的制备方法,并非是对可溶性聚酰亚胺固体的限制,市面上出售的或其他制备方法制备出的符合纯度要求的可溶性聚酰亚胺固体均可用于聚酰亚胺基体溶液的制备。
(四)制备聚酰胺-聚酰亚胺双链共混复合物
在所述聚酰亚胺基体液中缓慢加入至少一种引入氢键作用的聚合物单体,生成包含R
该结构为含酰胺键(-NH-CO-)的芳香聚酰胺分子链与聚酰亚胺分子链形成的双链结构,且所述双链的链间具有互穿的氢键网络结构。含酰胺键的芳香聚酰胺分子链的链刚度和线性度有助于聚酰亚胺分子链的取向和双链链间互穿网络结构的形成,链取向和双链链间互穿网络使分子刚性增加、运动能力减小。同时,由芳香聚酰胺分子链的有序氢键与聚酰亚胺分子链的亚胺环形成的链间互穿的氢键网络结构,能有效避免芳香聚酰胺分子间因有序氢键引起的聚合物链结晶,使芳香聚酰胺分子链呈现为分子水平分散的刚性聚酰胺聚合物。
通过控制含酰胺键的芳香聚酰胺分子链与聚酰亚胺分子链的摩尔比,可控制双链且链间具有互穿的氢键网络结构的占比。
具体的,所述的R
其中,各结构式中的R
各结构式中的R
以上结构式中~波浪线表示连接键,P为正整数。
所述的R
以上各结构式中的R
(五)制备聚酰胺-聚酰亚胺双链共混薄膜
将所述聚酰胺-聚酰亚胺双链共混聚合物涂覆到基板上,加热固化,得到所述聚酰胺-聚酰亚胺复合薄膜。
具体地,将所述聚酰胺-聚酰亚胺双链共混聚合物均匀的涂覆在玻璃基板上,在鼓风烘箱中60℃干燥1h,再在真空烘箱中采用梯度加热法加热至360℃除去极性非质子溶液和反应产生的盐酸,得到厚度为20~30um的聚酰胺-聚酰亚胺复合薄膜。
所述的梯度加热法具体的加热步骤为:第一梯度,加热至80℃并保温1h;第二梯度,加热至180℃并保温1h;第三梯度,加热至250℃并保温1h;第四梯度,加热至300℃并保温1h;第五梯度,加热至360℃并保温1h。通过该加热方法制备的薄膜致密性好,厚度均匀,缺陷少。
可通过控制含酰胺键的芳香聚酰胺分子链与聚酰亚胺分子链的摩尔比,调控由聚酰胺-聚酰亚胺复合物制备的薄膜的热膨胀系数、光学性能和力学性能,以满足不同柔性器件对超低热膨胀系数、高透光性、高耐热性及高耐有机溶剂性的个性化需求。
下面结合实施例和对比例对本发明的技术方案和技术效果作进一步说明。
需要说明的是,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
本实施例按照以下方法制备芳香聚酰胺与聚酰亚胺摩尔比为0.11:1的聚酰胺-聚酰亚胺复合薄膜。
准备原材料,其中,二胺单体选用2,2'-双(三氟甲基)-4,4'-二氨基联苯(TFMB);二酐单体选用六氟二酐(6FDA);极性非质子溶剂选用N,N-二甲基乙酰胺(DMAC);酰化剂选用乙酸酐与吡啶的体积比为7:3的混合溶液;引入氢键的聚合物单体采用对苯二甲酰氯(TC)。
(一)制备前驱体聚酰胺酸
S11将0.01mol的TFMB置于50mL的氮气保护的三口烧瓶中,随后向所述三口烧瓶中加入43.32mL的DMAC,使TFMB完全溶解于DMAC;
S12将0.01mol的6FDA逐步加入到所述三口烧瓶中,得到6FDA与TFMB的混合物,此时溶液固含量为15%;
S13将所述混合物在0℃下的氮气氛中连续搅拌12h,得到粘稠的前驱体聚酰胺酸。
(二)制备可溶性聚酰亚胺固体
S21将酰化试剂在室温下逐步加入到所述前驱体聚酰胺酸中,并在室温下连续剧烈搅拌24h,生成聚酰亚胺胶液,其中酰化试剂的用量满足酰化试剂中的乙酸酐与所述前驱体聚酰胺酸中的羧基的摩尔比等于5,即[Ac
S22用所述酰化试剂将所述聚酰亚胺胶液稀释成均相胶液,将稀释后的聚酰亚胺胶液缓慢倒入大量乙醇中静置沉降,收集沉降物并用新鲜乙醇反复洗涤、过滤,将经新鲜乙醇抽提的沉降物在120℃下真空干燥12h,得到可溶性聚酰亚胺固体。
(三)制备聚酰亚胺基体溶液
S31将所述可溶性聚酰亚胺固体溶解在无水的DMAC中,配置成含聚酰亚胺分子链的聚酰亚胺溶液,其中DMAC的用量满足所述聚酰亚胺溶液的固含量为10%;
S32将0.0011mol的TFMB加入到所述聚酰亚胺溶液中,使TFMB完全溶解于所述聚酰亚胺溶液,得到聚酰亚胺基体液。
(四)制备聚酰胺-聚酰亚胺双链共混复合物
S41将0.0011mol的TC逐步加入到所述聚酰亚胺基体胶液中,并在0℃下搅拌24h,得到含聚酰胺-聚酰亚胺双链共混聚合物的胶液。
(五)制备聚酰胺-聚酰亚胺双链共混薄膜
S51将所述聚酰胺-聚酰亚胺双链共混聚合物的胶液均匀的涂覆在玻璃基板上,在鼓风烘箱中60℃干燥1h;
S52再将涂覆有聚酰胺-聚酰亚胺双链共混聚合物的玻璃基板在真空烘箱中采用梯度加热法加热至360℃后保温1h,除去极性非质子溶液和反应产生的盐酸,得到厚度为20~30um、聚酰胺与聚酰亚胺摩尔比为0.11:1的聚酰胺-聚酰亚胺复合薄膜。
其中,梯度加热法具体的加热步骤为:第一梯度,加热至80℃并保温1h;第二梯度,加热至180℃并保温1h;第三梯度,加热至250℃并保温1h;第四梯度,加热至300℃并保温1h;第五梯度,加热至360℃并保温1h。
实施例2
本实施例按照以下方法制备聚酰胺与聚酰亚胺摩尔比为0.25:1的聚酰胺-聚酰亚胺复合薄膜。
本实施例的制备方法与实施例1基本相同,不同之处在于步骤S32和S41,在所述聚酰亚胺溶液中加入的二胺单体与引入氢键的聚合物单体的摩尔量不同,具体地为:
S32将0.0025mol的TFMB加入到所述聚酰亚胺溶液中,使TFMB完全溶解于所述聚酰亚胺溶液,得到聚酰亚胺基体液。
S41将0.0025mol的TC逐步加入到所述聚酰亚胺基体胶液中,并在0℃下搅拌24h,得到含聚酰胺-聚酰亚胺双链共混聚合物的胶液。
实施例3
本实施例按照以下方法制备聚酰胺与聚酰亚胺摩尔比为0.43:1的聚酰胺-聚酰亚胺复合薄膜。
本实施例的制备方法与实施例1基本相同,不同之处在于步骤S32和S41,在所述聚酰亚胺溶液中加入的二胺单体与引入氢键的聚合物单体的摩尔量不同,具体地为:
S32将0.0043mol的TFMB加入到所述聚酰亚胺溶液中,使TFMB完全溶解于所述聚酰亚胺溶液,得到聚酰亚胺基体液。
S41将0.0043mol的TC逐步加入到所述聚酰亚胺基体胶液中,并在0℃下搅拌24h,得到含聚酰胺-聚酰亚胺双链共混聚合物的胶液。
实施例4
本实施例按照以下方法制备聚酰胺与聚酰亚胺摩尔比为0.67:1的聚酰胺-聚酰亚胺复合薄膜。
本实施例的制备方法与实施例1基本相同,不同之处在于步骤S32和S41,在所述聚酰亚胺溶液中加入的二胺单体与引入氢键的聚合物单体的摩尔量不同,具体地为:
S32将0.0067mol的TFMB加入到所述聚酰亚胺溶液中,使TFMB完全溶解于所述聚酰亚胺溶液,得到聚酰亚胺基体液。
S41将0.0067mol的TC逐步加入到所述聚酰亚胺基体胶液中,并在0℃下搅拌24h,得到含聚酰胺-聚酰亚胺双链共混聚合物的胶液。
对比例
本对比例是没有混合增强项聚酰胺的聚酰亚胺薄膜的制备方法,具体地,包括以下步骤:
准备原材料,其中,二胺单体选用2,2'-双(三氟甲基)-4,4'-二氨基联苯(TFMB);二酐单体选用六氟二酐(6FDA);极性非质子溶剂选用N,N-二甲基乙酰胺(DMAC);酰化剂选用乙酸酐与吡啶的体积比为7:3的混合溶液。
(一)制备前驱体聚酰胺酸
S11将0.01mol的TFMB置于50mL的氮气保护的三口烧瓶中,随后向所述三口烧瓶中加入43.32mL的DMAC,使TFMB完全溶解于DMAC;
S12将0.01mol的6FDA逐步加入到所述三口烧瓶中,得到6FDA与TFMB的混合物,此时溶液固含量为15%;
S13将所述混合物在0℃下的氮气氛中连续搅拌12h,得到粘稠的前驱体聚酰胺酸。
(二)制备可溶性聚酰亚胺固体
S21将酰化试剂在室温下逐步加入到所述前驱体聚酰胺酸中,并在室温下连续剧烈搅拌24h,生成聚酰亚胺胶液,其中酰化试剂的用量满足酰化试剂中的乙酸酐与所述前驱体聚酰胺酸中的羧基的摩尔比等于5,即[Ac
S22用所述酰化试剂将所述聚酰亚胺胶液稀释成均相胶液,将稀释后的聚酰亚胺胶液缓慢倒入大量乙醇中静置沉降,收集沉降物并用新鲜乙醇反复洗涤、过滤,将经新鲜乙醇抽提的沉降物在120℃下真空干燥12h,得到可溶性聚酰亚胺固体。
(三)制备聚酰亚胺溶液
S31将所述可溶性聚酰亚胺固体溶解在无水的DMAC中,配置成含聚酰亚胺分子链的聚酰亚胺溶液,其中DMAC的用量满足所述聚酰亚胺溶液的固含量为10%;
(四)制备聚酰亚胺薄膜
S41'将所述聚酰亚胺溶液均匀的涂覆在玻璃基板上,在鼓风烘箱中60℃干燥1h;
S42'再将涂覆有聚酰亚胺溶液的玻璃基板在真空烘箱中采用梯度加热法加热至360℃后保温1h,除去极性非质子溶液,得到厚度为20~30um的聚酰亚胺薄膜。
其中,梯度加热法具体的加热步骤为:第一梯度,加热至80℃并保温1h;第二梯度,加热至180℃并保温1h;第三梯度,加热至250℃并保温1h;第四梯度,加热至300℃并保温1h;第五梯度,加热至360℃并保温1h。
结果测试分析
对实施例1至实施例4制得的聚酰胺-聚酰亚胺薄膜和对比例制得的聚酰亚胺薄膜进行性能测试分析。
测试环境条件:温度15℃~35℃,相对湿度40%~60%。
测试设备与测试条件:
1.热重(TGA)
测试设备:Perkinelmer PE Pyrisl TGA;
测试条件:样品质量为3~5mg,气体流速40mL·min
2.动态热机械分析(DMA)
测试设备:DMA Q800;
测试条件:薄膜样品,在薄膜拉伸模式下测试,测试频率为1Hz,在空气气氛中测试,升温速率为10℃·min
3.热机械衍射(TMA)
测试设备:TMA Q400;
测试条件:薄膜样品,在薄膜拉伸夹具上测试,测试频率为1Hz,在空气气氛中测试,升温速率为10℃·min
4.紫外-可见光光谱仪(UV)
测试设备:SHIMADZU U-3900型紫外-可见光光谱仪
测试条件:测试光谱为透过类型,扫描速率为300nm·min
5.凝胶渗透色谱(GPC):
测试设备:Waters公司的Breeze GP系统;
测试条件:测试溶剂为DMF,样品浓度为1-2mg·mL
测试结果如下:
1光学性能
表1
/>
其中:T
由表1所示,通过原位聚合将芳香聚酰胺引入聚酰亚胺中,其光学性能得到了基本的维持。
2热性能
表2
其中:T1%、T5%分别表示在氮气氛围下热失重分别为1%和5%的温度;T
由表2所示,通过原位聚合将聚酰胺引入到聚酰亚胺中,其热性能得到了很好的维持。随着芳香聚酰胺含量的增加,CTE明显降低,当芳香聚酰胺含量与聚酰亚胺含量比达到0.67:1时,CTE值为0.8ppm·K
通过以上各项数据表面:本发明制得的聚酰胺-聚酰亚胺复合物薄膜,在保持聚酰亚胺薄膜的特有的光学透明度和热性能的同时,能有效改善聚酰亚胺薄膜尺寸稳定性和机械性能较差的问题。通过本发明的制备方法制备的聚酰胺-聚酰亚胺复合物薄膜,具有更低的热膨胀系数和更高的玻璃化转变温度,拥有优异的尺寸热稳定性。
本发明设计的采用原位聚合将增强相芳香聚酰胺分子链均匀的分散到聚酰亚胺基体中的方法,能得到大分子量的聚酰亚胺分子链和芳香聚酰胺分子链形成的双链互穿网络结构;双链间的相互作用使界面空隙率最小化,能有效避免光散射,实现薄膜的高光透过率;双链间的互穿氢键网络结构使分子链刚性增加、相对运动能力减小、分子间距减小、链段紧密堆砌,能有效提高薄膜的热尺寸稳定性,低的热膨胀系数使薄膜在器件加工过程中不易卷曲或剥离;高的玻璃化转变温度保证薄膜在高温气氛中加工时不发生尺寸变化甚至熔化。
本发明并不局限于上述实施方式,如果对本发明的各种改动或变形不脱离本发明的精神和范围,倘若这些改动和变形属于本发明的权利要求和等同技术范围之内,则本发明也意图包含这些改动和变动。
- 一种低粘度聚酰亚胺共聚热固性树脂及其制备方法和应用
- 一种聚酰亚胺纤维紧密纺纱及其制备方法和应用
- 一种3D打印用光固化聚酰亚胺墨水的制备方法及其应用
- 一种软质聚酰亚胺泡沫、其制备方法及其应用
- 一种具有光致变色性能的聚酰亚胺及其制备方法和应用
- 一种聚酰胺酸溶液及其制备方法、聚酰亚胺薄膜及其制备方法和应用
- 一种聚酰胺酸溶液及其制备方法、聚酰亚胺薄膜及其制备方法和应用