低溶性药物的连续热熔融制粒方法
文献发布时间:2024-04-18 19:58:21
本发明涉及一种在熔融制粒方法中使聚合物负载活性药物成分的方法和所制备的产品。更具体地讲,本发明涉及制备颗粒的方法,所述颗粒包含至少一种活性药物成分和聚乙烯醇。
发明背景
为了在药物制剂中实现更一致的活性药物成分剂量率,当活性药物成分作为均匀分散体或溶液存在于载体时是有用的。尤其是,低溶性药物物质的溶解度提高是用于无定形固体分散体的重要应用。
固体分散体被定义为一种或多种活性药物成分在惰性固体基质中的分散体,并可概括分类为包含结晶态或无定形态药物物质的那些分散体[Chiou W.L.,RiegelmanS.Pharmaceutical applications of Solid dispersion systems;J.Pharm Sci.1971,60(9),1281–1301]。包含结晶态药物活性成分的固体分散体通过简单地降低表面张力、减少附聚和改善活性物质的润湿性提供溶解度的提高[Sinswat P.,et al.;Stabilizerchoice for rapid dissolving high potency itraconazole particles formed byevaporative precipitation into aqueous solution;Int.J.of Pharmaceutics,(2005)302;113–124]。然而结晶系统比无定形对应物在热力学上更稳定,结晶结构必须在溶解过程中断开,这需要能量。称为无定形固溶体的包含活性药物成分(这指在分子水平上溶解的药物)的固体分散体,可导致溶解速率和过饱和程度显著提高[DiNunzio J.C.et al.IIIAmorphous compositions using concentration enhancing polymers for improvedbioavailability of itraconazole;Molecular Pharmaceutics(2008);5(6):968–980]。虽然这些系统有若干优点,但由于分子流动性和药物重结晶的倾向导致的物理不稳定性可能是成问题的。具有高玻璃化转变温度的聚合物载体似乎很适合通过限制分子流动性来稳定这些系统。
因此,固体分散体可通过多种方法产生,包括但不限于喷雾干燥、熔融挤出或热动力学配混。
热熔融挤出(HME)最近在用于制备通过挤出加工的包含活性药物成分的制剂的制药工业中获得了认可。HME已作为制药技术引入,并已成为众所周知的方法,具有益处如连续和有效的加工、有限的方法步骤数、无溶剂方法等。在热熔融挤出过程中,活性药物成分、热塑性赋形剂和其它功能性加工助剂的混合物在挤出机内被加热和软化或熔融,并通过喷嘴挤出成不同的形式。
固体分散体也可通过制粒技术产生。制粒是一种使一级药物和赋形剂颗粒附聚成较大二级颗粒或粒的成熟药物加工技术。在制药工业中已建立了许多种湿制粒和干制粒技术。在连续制粒领域,双螺杆制粒方法是最有前途的技术。连续双螺杆湿制粒(TSWG)已经常用于避免流动和压缩问题。这里的缺点是通过湿加工的稳定性和降解问题以及相关干燥步骤的适当控制。
双螺杆熔融制粒(TSMG)可以提供湿制粒的有趣替代方案。通常,通过软化或熔化粘合剂而不是制粒液体引发附聚,使这项技术极其适用于湿度敏感的药物。熔融制粒被认为是一种尺寸放大方法,其中用加入在相对较低的温度(通常在约60℃)熔融或软化的粘合剂实现固体颗粒的附聚。不同的技术已经成熟,包括喷雾冷凝和翻滚熔融制粒。
从(热)熔融挤出技术,已知聚合物应具有合适的性质,例如热塑性、合适的玻璃化转变温度或熔点、在所需加工温度的热稳定性、与活性药物成分没有意外的化学相互作用等。
发明目的
常见的熔融制粒技术需要使用可熔粘合剂。那些可熔粘合剂通常是在相对较低温度(50℃至90℃)熔融或软化的低熔点物质,例如低熔点蜡或低熔点聚合物。可熔粘合剂用于在制粒方法期间实现固体颗粒的附聚。通常,加工温度设定在高于聚合粘合剂的Tm或Tg但低于药物物质的Tm。这将药物保持在其结晶态,以最大限度地减小任何物理化学变化(Kittikunakorn N,Liu T,Zhang F.Twin-screw melt granulation:Current progressand challenges,International Journal of Pharmaceutics,2020;588:119670)。已有人描述了不同类型的亲水性和疏水性粘合剂。通常,熔融粘合剂在固体颗粒表面上的分布在熔融制粒过程中由混合和紧压二者来驱动。
也经常使用脂类。然而,低熔点粘合剂承担着处理和储存附聚物期间也发生粘合剂熔融或软化的风险。另一个问题是合适聚合物的数量有限。
因此,本发明的一个目的是提供一种熔融制粒方法,以获得聚合物中或聚合物上API的无定形固体分散体。本发明的另一个目的是提供一种组合物,组合物可以在不需要粘合剂下用于熔融制粒方法。另一个目的是提供一种组合物,其中熔融制粒方法得到聚合物中或聚合物上API的无定形固溶体或分散体,而不需要粘合剂。另一个目的是提供制粒方法参数,以实现上述内容。
此外,本发明的一个目的是提供一种挤出方法,其中产品具有有益的性质,例如无定形化或溶解的程度。
发明内容
在常见的熔融制粒方法中,用粘合剂形成结晶药物物质的附聚物。通常低熔点聚合物是优选的,因为在大多数情况下目的只是附聚,以增加粒度。
惊讶地发现,可用熔融制粒方法使聚合物负载无定形形式的活性药物成分(API)。通过在熔融制粒方法期间用聚乙烯醇(PVA)作为聚合物,聚合物可作为稳定无定形形式的API的基础。此外,令人惊讶地发现,当将API加热到高于其熔点时,获得关于无定形形式的稳定化和API溶解的特别优选的结果。在冷却步骤期间,液体药物物质在聚合物内或在聚合物表面上固化,并保持其无定形态,以形成无定形固溶体或分散体。
此外,还发现所得颗粒具有与由HME制备的颗粒相比有益的性质。
还发现双螺杆熔融制粒(TSMG)特别适合作为制粒方法。
具体实施方式
本发明涉及一种在熔融制粒方法中使聚合物负载活性药物成分的方法,所述方法包括以下步骤:
a)在挤出机的加热螺杆机筒中捏和包含至少一种活性药物成分和聚乙烯醇的混合物,其中沿螺杆机筒长度的至少一个区域中的温度高于至少一种活性药物成分的熔融温度,且低于聚乙烯醇的分解温度,以形成捏和混合物,和
b)通过出口输送捏和混合物。
“活性药物成分”或“API”可以一种或多种药学上可接受的盐、酯、衍生物、类似物、前药及其溶剂化物的形式发现。如本文所用“药学上可接受的盐”理解为指由酸和碱相互作用形成的化合物,酸的氢原子由碱的阳离子取代。
术语“聚乙烯醇”或“PVA”是指具有理想化式[CH
典型的PVA命名法指示了聚合物4%溶液在20℃的粘度和水解度。例如,PVA 3-83为具有3mPas粘度的83%水解的PVA等级,即具有83%的乙烯醇重复单元和17%的乙酸乙烯酯重复单元。技术人员认识到,83%的水解等级和3mPas的粘度包括根据常用舍入方法计算的82.50%至83.49%的水解等级和计算的2.50mPas至3.49mPas%的粘度。根据本发明的粘度如USP 39中专论“聚乙烯醇”下所述用方法粘度-旋转法<912>测量。根据本发明的水解度如USP 39中专论“聚乙烯醇”的“水解度”下所述测量。
随着水解度增加,聚合物在水性介质中的溶解度增加,而且聚合物的结晶度增加。除此之外,玻璃化转变温度和熔融温度也依其水解度、分子量和水含量而变。例如,干燥损失≤5.0%的PVA 4-88的熔融温度约为170℃,玻璃化转变温度约为40至45℃,分解温度>250℃(Technical Information,
聚乙烯醇可溶于水,但几乎不溶于几乎所有的有机溶剂,在某些情况下不包括乙醇。聚合物的这个方面使得当药物在水性介质中也具有有限的溶解度时,非常难以通过喷雾干燥形成无定形和固体分散体。
根据本发明的聚乙烯醇可包括任何PVA等级。
在一个实施方案中,聚乙烯醇由不同分子量和不同水解等级的一种或多种等级的PVA组成。
在一个实施方案中,聚乙烯醇具有72%至90%、具体地讲74%至88%、具体地讲80%至90%的水解度和4%溶液在20℃下2mPas至40mPas、具体地讲3mPas至18mPas的粘度。
在一个实施方案中,聚乙烯醇选自PVA 3-80、PVA 3-81、PVA 3-82、PVA 3-83、PVA3-85、PVA 3-88、PVA 3-98、PVA 4-88、PVA 4-98、PVA 5-74、PVA 5-82、PVA 6-88、PVA 6-98、PVA 8-88、PVA 10-98、PVAPVA 13-88、PVA 15-99、PVA 18-88、PVA 20-98、PVA 23-88、PVA26-80、PVA 26-88、PVA 28-99、PVA 30-98、PVA 30-92、PVA 32-88和PVA 40-88。
在另一个实施方案中,聚乙烯醇选自PVA 3-80、PVA 3-81、PVA 3-82、PVA 3-83、PVA 3-88、PVA 4-88、PVA 5-74、PVA 5-88、PVA 8-88和PVA 18-88。
在另一个实施方案中,聚乙烯醇选自PVA 3-80、PVA 3-81、PVA 3-82、PVA 3-83、PVA 4-88和PVA 18-88。
在另一个实施方案中,聚乙烯醇为PVA 4-88。在另一个实施方案中,聚乙烯醇为PVA 3-82。
在另一个实施方案中,低温研磨聚乙烯醇。优选将PVA低温研磨到适用于熔融制粒方法的粒度。示例性市售PVA为
术语“熔融温度”是指物质从固态变成液态的温度。在熔融温度,固相和液相平衡存在。物质的熔融温度依赖压力,根据本发明,规定熔点在1个大气压的压力。PVA等级的熔融温度依赖各自PVA等级的水解度和粘度。PVA的熔融温度一般在180至220℃的范围内。
术语“玻璃化转变温度”是指在无定形材料中,或在半结晶材料内的无定形区域中,随着温度升高,从硬且相对脆的“玻璃”态逐渐且可逆地转变成粘性或橡胶态。物质的玻璃化转变温度依赖压力,根据本发明,规定玻璃化转变温度在1个大气压的压力。PVA等级的玻璃化转变温度依赖各自PVA等级的水解度和粘度。PVA的玻璃化转变温度一般在40至80℃的范围内。
术语“分解温度”是指引起其中热正在断裂化学键的化学分解的温度。物质的分解温度依赖压力,根据本发明,规定分解温度在1个大气压的压力。PVA等级的分解温度依赖各自PVA等级的水解度和粘度。PVA的分解温度一般在250℃的温度开始。
与使用低熔点粘合剂用于固体颗粒附聚的常见热熔融制粒技术不同,此处所述的方法在高于活性药物成分熔点和高于PVA玻璃化转变温度的温度进行。实验显示,PVA由于其半结晶性质是一种非常有前途的载体。PVA可以稳定固体分散体中活性药物成分的无定形相。
在文献中,使用较低熔点粘合剂是优选的,因为较高熔点的粘合剂需要高的熔融温度,并且可能导致不稳定性问题,尤其对于热不稳定材料,如热不稳定API(Meltgranulation:An alternative to traditional granulation techniques;March 2013;Indian Drugs 50(3):5-13)。根据本发明,选择使API在制粒方法期间熔融的温度。惊讶地发现,可用熔融的API本身作为粘合剂,并与移动化的PVA紧密接触。在制粒系统内紧密相互结合确保了PVA和API之间的强相互作用,并引起API在聚合物中均匀分布。
根据本发明,用于在熔融制粒方法中获得API的无定形固体分散体的最低工作温度是在沿螺杆机筒长度的至少一个区域中高于API熔融温度的温度。最高工作温度为PVA的分解温度。PVA的玻璃化转变温度根据聚合和水解程度在40℃和80℃之间变化。多数PVA等级的分解在约250℃开始。因此,根据本发明的方法可用于熔点在40℃和250℃之间的API。获得PVA聚合物中API的无定形固体分散体的典型工作温度为140℃至230℃,优选170℃至210℃,更优选180℃至200℃。
根据本发明的双螺杆熔融制粒与其它加工技术(如热熔融挤出(HME))相比提供了一个主要的益处,热熔融挤出(HME)迫使塑化熔体通过在挤出机筒末端连接的模头。在根据本发明的方法中,不使混合物通过模头,而是通过出口,出口是通向PVA颗粒的开口,其中HME通向PVA粒料。颗粒中的活性药物成分更好地稳定化,和/或显示出改善的溶解度分布,并且可更容易地加工成最终剂型(片剂)。
在一个实施方案中,沿螺杆机筒长度的至少一个区域中的温度高于至少一种活性药物成分的熔融温度,且低于聚乙烯醇的玻璃化转变温度和分解温度。
如果温度高于PVA的熔融温度,则通过出口输送的捏和混合物处于熔融态。因此,在步骤b)将捏和混合物输送通过出口,以获得熔融/熔化混合物。
在一个实施方案中,沿螺杆机筒长度的至少一个区域中的温度高于至少一种活性药物成分的熔融温度,且低于聚乙烯醇的熔融温度,优选在高于至少一种活性药物成分的熔融温度且在聚乙烯醇的玻璃化转变温度和熔融温度之间的温度。在那个实施方案中,通过出口输送的捏和混合物为颗粒形式。因此,在步骤b)将捏和混合物输送通过出口,以获得颗粒。
在另一个实施方案中,沿螺杆机筒长度的至少一个区域中的温度在40℃至250℃之间,优选140℃至230℃,更优选170℃至210℃,最优选180℃至200℃。
在另一个实施方案中,如上所述的温度在沿螺杆机筒长度的所有区域中是相同的。
术语“熔融制粒方法”或“HMG”一般指尺寸放大方法,其中用加入熔融或软化的粘合剂实现制剂中固体颗粒的附聚。方法利用了当它们在软化或熔化态时作为制粒剂有效的材料。在制药工业中,可用这种方法制备快速释放或缓释剂型。粘合剂或可熔粘合剂通常为在相对较低温度(50℃至90℃)熔融或软化的低熔点物质,例如低熔点蜡或低熔点聚合物。可用可熔粘合剂在制粒方法期间实现固体颗粒的附聚。与HME相反,熔融制粒方法中的混合物通过为开口但不是喷嘴或模头的出口输送。这意味着以不会对捏和混合物施加压力的方式形成所需尺寸的出口。这与模头相反,模头会以压力能量为代价来导致提高流体的速度。
由HMG得到的产品与由HME制备的那些产品不同。HME颗粒具有角形形状和相对平坦的表面,其中HMG颗粒具有更圆形的形状与更不平坦的表面,这可通过SEM测量看到。HMG颗粒具有较低的比表面积。意外的是,在可比的粒度分布下(例如HME3-750μm和HMG4-350rpm),由HMG制备的颗粒显示出更快的API溶解,尽管它们具有较低的比表面积。令人惊讶的是,由HMG制备的颗粒的溶解随着粒度增加而更快,这可在图13中看到。这与由HME制备的颗粒的情况(图12)不一样。
根据本发明不需要额外的粘合剂。附聚的程度取决于螺杆机筒的温度。在上述方法的温度范围内(“高于至少一种活性药物成分的熔点且在聚乙烯醇的玻璃化转变温度和分解温度之间”),制粒方法期间的附聚程度可通过选择特定温度来调节。在低于PVA熔融温度的温度,可检测到低程度的附聚。随着温度升高,附聚的程度增加。惊讶地发现,另外地,熔化和液化的API可充当粘合剂,以增加附聚。
与粒度和附聚程度无关,已令人惊讶地发现,在上述条件下,API以无定形形式负载在PVA颗粒上,并以该形式稳定化。
无定形固体分散体可任选包含其它药学上可接受的组分。
如本文所用短语“药学上可接受的”是指一般在对人施用时不产生过敏或类似不良反应的所有化合物,如溶剂、分散介质、赋形剂、载体、包衣料、活性剂、等渗和吸收延迟剂等。在药物组合物中使用这样的介质和药剂在本领域是熟知的。
在一个实施方案中,颗粒中的活性药物成分以无定形形式分散于聚乙烯醇内和/或聚乙烯醇的表面上。
术语“以无定形形式分散”是指无定形API在聚合物内或在聚合物表面上分散。优选无定形API以分子分散态分布在聚合物表面上。溶解后,包含无定形固体分散体的制剂在水性介质中可以达到比结晶API更高的溶解度。
在一个实施方案中,在本发明药物组合物中包括的API具有治疗有效的足够量。对于给定的API,治疗有效量通常是本领域技术人员已知的或容易获得的。通常,API可以在1:99至90:10、优选5:95至60:40、最优选10:90至30:70范围的API与PVA重量比存在于药物组合物中。
术语“挤出机”是指包含一个或多个旋转螺杆的机筒,螺杆将材料沿着机筒向下输送。那些挤出机包括(i)材料进入机筒通过的开口,开口可具有料斗,料斗填充有待挤出或由一个或多个外部进料器以受控方式连续供应的材料;(ii)传送(过程)区段,该区段包括机筒和螺杆,其输送材料并且在适当时将材料混合,和(iii)任选的下游辅助设备,用于冷却、切割、分类和/或收集成品。根据本发明,合适的挤出机为单螺杆挤出机、双螺杆挤出机或行星辊挤出机。双螺杆挤出机是优选的。
在一个实施方案中,熔融制粒方法为双螺杆熔融制粒法。
术语“双螺杆熔融制粒”(TSMG)是指特定形式的熔融制粒方法。在双螺杆熔融制粒中,使用双螺杆,双螺杆由安装在封闭机筒中花键轴上的两个相互啮合、共同旋转螺杆组成。由于宽范围的螺杆和机筒设计,可根据方法要求设置各种螺杆型材和方法功能。双螺杆能够确保以高度的灵活性输送、压缩、混合、蒸煮(cooking)、剪切、加热、冷却、泵送、成形。
如本文所用术语“热熔融挤出”或“HME”是指其中活性药物成分、热塑性赋形剂和其它功能性加工助剂在挤出机内被加热和软化或熔融,并通过至少一个喷嘴或模头挤出成不同形式的方法。
根据本发明的至少一种活性药物成分(API)为生物活性剂,它可以为一种或多种药学上可接受的盐、酯、衍生物、类似物、前药及其溶剂化物的形式。药物组合物可包含一种或多种API。
根据生物制药分类系统(BCS)第2类和第4类按照低溶解度的定义,如本文所用术语“难溶性API”、“水难溶性API”和“亲脂性API”是指API的溶解度使得最高治疗剂量的待施予个体的特定API不能溶于250ml 1至8pH范围的水性介质。分别落入BCS第2或第4类的API为本领域的技术人员所熟知的。BCS第2类的难溶性API的典型实例为伊曲康唑(ITZ)。
在一个实施方案中,活性药物成分为难溶性API。
在方法结束,优选以颗粒形式排出产物。这些颗粒可很容易地用于进一步的加工步骤,如研磨、胶囊填充或利用或不利用添加额外的赋形剂直接压缩。颗粒的粒度可通过方法参数变化来调整。
如本文所用术语“颗粒”是指大分子尺寸的主要球形、角形、近球形或近角形结构,优选具有20-2500μm之间、更优选50-2000μm之间、最优选100-1500μm之间的平均粒度。“平均粒度”定义为取样粉末或颗粒的质量(颗粒)的50%具有较小直径的等效直径。对于粒度分布的测量,不同的方法是可用的。根据本发明,通过动态图像分析(ISO 13322-2)测量粒度分布。在一个特定实施方案中,用来自Retsch GmbH的Camsizer X2测量粒度分布,优选在测定期间启动CCD-B和CCD-Z两者的相机系统,用于分散的气压设定到50kPa,狭缝宽度设定到4mm。
在此通过熔融制粒进行的制粒方法的概念不仅限于双螺杆熔融制粒。如果API和聚合物可以在适当的温度条件下加工,也可以采用分批进行的其它热加工技术。
为了说明本发明,以所述方式,将伊曲康唑(ITZ)作为低溶模型活性物质加工,并使用公开的熔融制粒方法与聚乙烯醇加工。要强调的是,本发明不限于ITZ或低溶性API。如上所述,可利用熔融温度在40℃和250℃之间的所有API进行这种方法。
本发明进一步涉及用上述方法生产PVA中至少一种活性药物成分的无定形固体分散体的方法。本发明进一步涉及用上述方法在PVA内分散无定形活性成分的方法。
本发明进一步涉及可通过上述方法获得的颗粒。通过上述方法获得的颗粒可直接填入囊袋或胶囊,或进一步加工成片剂、胶囊剂或多颗粒系统。
通常,颗粒至少包含API和PVA。它们可任选包含其它药学上可接受的组分。
至少一种API和PVA可以在1:99至90:10、优选5:95至60:40、最优选10:90至30:70范围的API与PVA重量比存在于颗粒中。
颗粒优选包含至少50%(重量/重量),更优选至少80%,最优选至少90%无定形形式的API。可任选将颗粒进一步研磨到限定粒度。优选将颗粒研磨到50μm至300μm之间的平均粒度。
本发明进一步涉及可通过上述方法获得的片剂。
虽然以下详细讨论了本发明的各种实施方案的制造和使用,但应理解,本发明提供了比此处详述更适用的发明概念。本文讨论的具体实施方案仅仅说明了制造和使用本发明的具体方式,而不限定本发明的范围。
本文未定义的词语具有与本发明相关的领域的普通技术人员通常理解的含义。诸如“一”、“一种”和“该”之类的词语不是仅旨在指单数实体,而是包括一般类别,其具体实例可用于说明。本文中的术语用于描述本发明的具体实施方案,但除了权利要求中概述的外,它们的使用不限定本发明。
实施例:
I.
1.
将结晶伊曲康唑和PVA 4-88(
螺杆的转速设定到300rpm。投料速率保持恒定于150g/h。单独加热区域的温度分布显示于表1中。
表1:显示在不同目标温度熔融制粒过程中挤出机的不同加热区域的温度分布。
2.
用粉末衍射仪测量结晶伊曲康唑和在实施例1中得到的颗粒。在装配有40ml容器的IKA Tubemill 100中以25000rpm研磨实施例1的颗粒20秒,并通过250μl筛过筛。
利用Rigaku Miniflex 600用以下设定测量PXRD:
在40kV,15mA产生X射线束。使用D/teX Ultra2检测器。扫描速度/持续时间设定到5°/min,步宽为0.02°。扫描范围设定到3.0-50.0°。
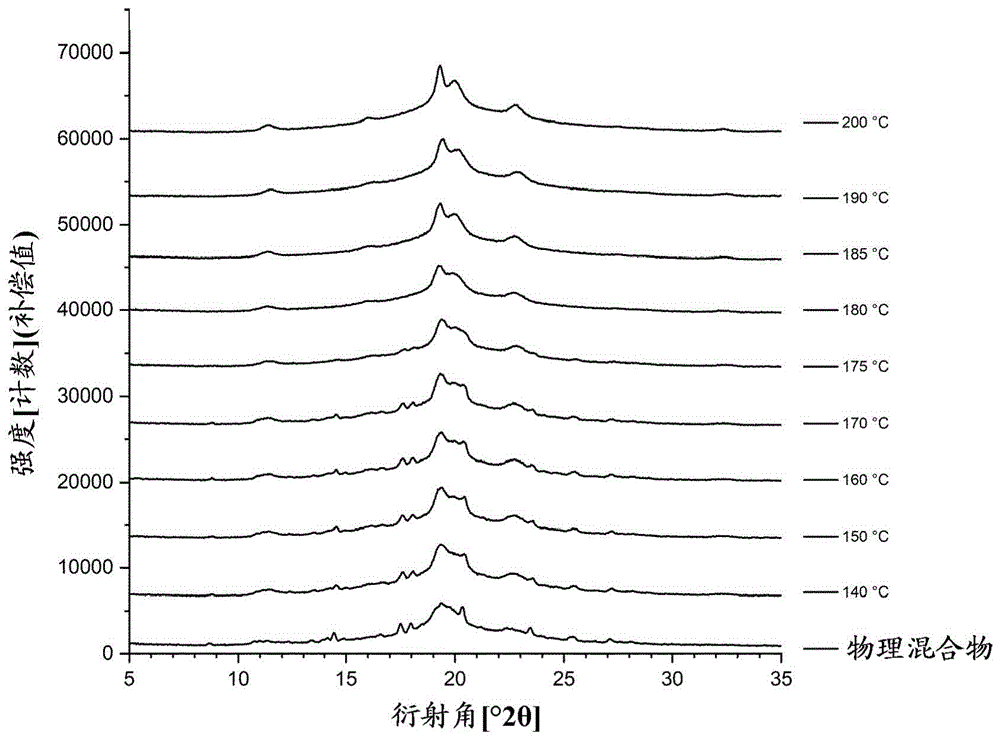
图1显示在表1中所示不同加工温度得到的不同热熔融颗粒的X射线衍射图。
图2显示用于实施例1的双螺杆熔融制粒方法的结晶伊曲康唑的X射线衍射图。
可在图1中看到,结晶伊曲康唑和PVA的物理混合物仍然含有结晶图案,例如在约14和21的衍射角的峰,其可清楚地与结晶伊曲康唑联系在一起(见图2)。也可在样品观察到同样的图案,直至约170℃的加工温度。在高于170℃的温度,所有药物物质似乎都转化成其无定形形式。伊曲康唑的熔融温度为166.2℃。因此,实验表明,需要高于熔点的温度来获得负载有无定形伊曲康唑的PVA颗粒。
3.
为了测量溶解度,直接使用如实施例1中获得的颗粒,而不进一步加工。每个温度设定使用3个样品。称重500mg 10%ITZ颗粒(Mettler Toledo Delta Range XP105),等于50mg ITZ API。
使用来自Analytik Jena的具有在线光度计Specord 200+的Sotax AT7 smart进行溶解。用37℃±0.5的900ml SGF.sp(20g NaCl,800ml0.1M HCl ad 10.0L VE-水)作为介质。使用以下设定:旋转速度75rpm;桨法;预过滤器:玻璃微纤维过滤器GE Whatmann GF/D直径25mm,5mm HELMA流通比色皿,采样点:5、20、35、50、60、120Min。
图3显示与结晶形式伊曲康唑相比,来自实施例1中获得的颗粒的伊曲康唑的溶解度分布曲线。
图3表明结晶伊曲康唑(线1)在释放介质中显示出非常低的溶解度。在140℃至170℃之间温度用聚乙烯醇制粒增加了化合物的溶解度(线2-6)。在175℃溶解度突然增加。随着较高温度,溶解度进一步提高。对180℃至190℃的温度观察到立即释放(线8-10)。随着温度进一步升高超过PCA的熔融温度(线11;200℃),药物释放速率延迟。
4.
为了测量粒度分布,直接使用1至3g如实施例1中获得的颗粒,而不进一步加工。使用来自Retsch GmbH的Camsizer X2测量粒度。在测定过程中启动相机系统CCD-B和CCD-Z二者。用于分散的气压设定到50kPa。狭缝宽度设定到4mm。体积百分比的累积分布在表2中给出。
表2:累积粒度分布(基于体积的通过百分比)。
II.热熔融挤出与热熔融制粒比较
1. 热熔融挤出(HME)
在混合容器中称重540.15g Parteck MXP和60.0g伊曲康唑,并在管式混合器中混合5分钟。
然后将聚合物/API混合物(比率:90/10)填入重力双螺杆进料器(Thermo Fisherscientific,Karlsruhe,Germany),并测量最大投料速率。最大投料速率:0.885kg/h。
用Pharma 11双螺杆挤出机(Thermo Fisher scientific,Karlsruhe,Germany)进行挤出,螺杆速度为250rpm,进料速率为0.15kg/h。扭矩为最大可能扭矩(最大扭矩=12Nm)的12%。
安装了2.0mm直径的圆孔喷嘴,也安装了排气口。
获得的白色、不透明纤丝由传送带(Brabender GmbH&Co.KG.,Duisburg,Germany)输送。传送带速度设定到1,49。在室温进行纤丝的冷却(Colling)。然后通过造粒机(Brabender GmbH&Co.KG.,Duisburg,Germany)切割纤丝,并收集白色颗粒。
热熔融挤出(利用喷嘴)的过程分布显示于图14中。
研磨:
用液氮冷冻来自实验的三批颗粒,每批60g。然后用ZM200超离心磨机(RETSCHGmbH,Haan,Germany)在以下条件研磨冷冻颗粒:
·18000rpm
·12齿转子,
·第一批利用350μm距筛(HME1-350μm批)
·第二批利用500μm距筛(HME1-500μm批)
·第三批利用750μm距筛(HME1-750μm批)
·带有旋风分离器的盒子
2.
在混合容器中称重450.0g Parteck MXP和50.1g伊曲康唑,并在管式混合器中混合5分钟。
然后将聚合物/API混合物(比率:90/10)填入重力双螺杆进料器(Thermo Fisherscientific,Karlsruhe,Germany),并测量最大投料速率。最大投料速率:0.705kg/h。
用装配有双螺杆制粒套件的Pharma 11双螺杆挤出机(Thermo Fisherscientific,Karlsruhe,Germany)进行热熔融制粒。
在190℃在200rpm、250rpm、300rpm、350rpm和400rpm的螺杆速度进行制粒。在指定rpm收集颗粒15分钟。在每个新的设定点后,在收集新批次之前舍弃颗粒另外10分钟。
热熔融制粒的过程分布显示于图15中。
3.
样品制备:
在用导电双面胶带覆盖的铝样品架上制备少量粉末。通过压缩空气或波纹管去除未粘附的颗粒,以防止松散的颗粒污染SEM的高真空室。为了避免静电充电,在测量前用~10nm铂涂覆样品(如果颗粒稍湿,建议在溅射前在溅射系统中在10-2bar干燥)。然后将制备的样品转移到SEM中,并在高真空下测量。
SEM仪器:
ZEISS Supra 35/LEO 1530,场发射阴极,分辨率高达2nm,高真空,放大倍数20x至500.000x,电压0.1kV至30kV,Inlens检测器,Everhart-Thornley检测器,4象限BSE检测器。图4至图11显示来自上述批次的颗粒的SEM图像。
图4:500×放大倍数的SEM图像HMG1-200rpm
图5:500×放大倍数的SEM图像HMG2-250rpm
图6:500×放大倍数的SEM图像HMG3-300rpm
图7:500×放大倍数的SEM图像HMG4-350rpm
图8:500×放大倍数的SEM图像HMG5-400rpm
图9:500×放大倍数的SEM图像HME1-350μm
图10:500×放大倍数的SEM图像HME2-500μm
图11:500×放大倍数的SEM图像HME3-750μm
4.
样品制备:使用未研磨样品:n=3(每粒)
称重500mg 10%ITZ颗粒等于50mg ITZ API(使用天平:Mettler Toledo DeltaRange XP105)
使用来自Analytik Jena的具有在线光度计Specord 200+的Sotax AT7 smart进行溶解
介质:37℃±0.5的900ml SGF.sp(20g NaCl,800ml 0.1M HCl ad10,0L VE-水)
旋转速度75rpm;桨法;预过滤器:玻璃微纤维过滤器GE Whatmann GF/D直径25mm
5mm HELMA流过比色皿
采样点:5,20,35,50,60,120Min
图12显示利用50mg API的不同批次的PVA4-88颗粒在900ml SGF、75rpm、桨法的溶解度。图13显示利用50mg API的批次HME3-750μm和HMG4-350rpm在900ml SGF、75rpm、桨法的溶解度比较。
可以看到,HMG4-350rpm与HME3-750μm相比具有更快的溶解。两个批次具有可比的粒度分布(见实施例II 8.)。一般而言,具有更高粒度的HMG颗粒显示出更快的溶解,对于HME颗粒这是相反的。
5.
在装配有40ml容器的IKA Tubemill 100中以25000rpm研磨颗粒20秒。通过250μl筛过筛。
SI-低背景样品架
PXRD方法:
·Rigaku Miniflex 600
·X射线40kV,15mA
·Goniometer MiniFlex 600
·附件ASC-8
·过滤器K-β(x1.5)
·检测器D/teX Ultra2
·扫描模式连续
·扫描速度/持续时间5.0000°/min
·步宽0.0200°
·扫描轴θ/2-θ
·扫描范围3.0000-50.0000°
·入射狭缝0.625°
·长度限制狭缝10.0mm
·接收狭缝#1 13.0mm(开放)
·接收狭缝#2 13.0mm(开放)
6.
将样品放入凹槽(1-3g)
为样品建立或分配相应的方法
开始测定
Camsizer方法:
·漏斗位置[mm]:5
·凹槽幅度[mm]:60
·狭缝宽度[mm]:4
·分散压力[kPa]:50
·速度调节
·尺寸限定:xc_min
·筛尺寸:Pharm.Eur
·参数:累积分布,球形度,幅度/长度,对称
·相机:CCD-基本/CCD-变焦
·帧速率:100%(1:1)
在5000张相片后完成测量
7.
通过气体吸附-BET法测量比表面积。对应于DIN ISO 9277:2014-01和ISO 9277:2010(E)进行测量。
样品的脱气和测量由来自Micromeritics Instrument Cooperation的“ASAP2420”仪器来进行,仪器使用静态-体积原理。
使用1.6g至4.5g样品量。将样品在40℃真空下干燥并脱气20小时。用氪(分子横截面积:0.2100nm
对于检查设备监测,使用两种参比材料:氧化铝(比表面积:0.22m
表5:比表面积结果。
8.
通过对应于ISO 13320:2020(E)的激光衍射光谱法测量粒度分布。
使用来自Malvern Panalytical Ltd.的具有干分散单元“Scirocco2000”的激光衍射光谱仪“Mastersizer 2000”。
分析约1.5g样品量。使用75%的进料率、6mm的间隙尺寸和3bar的气压,使样品分散于空气(折射率为1)。将遮光率范围设定到0.1%至10%。使用带有10个球的筛子(直径2mm)。
用具有通用目的分析模型的Fraunhofer模型评价光衍射图。表6显示了所述批次的粒度分布。HME3-750μm和HMG4-350rpm具有可比的粒度分布。
表6:粒度分布结果。
- 一种控缓释剂药用辅料的热熔融制粒方法
- 一种控缓释剂药用辅料的热熔融制粒方法