氘代醋酸甲地孕酮的合成方法及其应用
文献发布时间:2024-04-18 19:58:26
技术领域
本发明属于稳定性同位素标记化合物生产领域;更具体地,本发明涉及一种氘代醋酸甲地孕酮的合成方法及其应用。
背景技术
醋酸甲地孕酮是一种合成孕酮类药物,是孕酮的代表性衍生物之一。它被广泛用于妇科和避孕领域,可用于治疗月经不调、子宫内膜异位症、子宫肌瘤、乳腺癌等疾病,并可用于口服避孕药和紧急避孕药。
氘标记的醋酸甲地孕酮对于未标记的醋酸甲地孕酮在医学研究领域,特别是在药理学、药代动力学和生物合成途径的研究中有着极其重要的意义。但是,目前在醋酸甲地孕酮同位素标记这一方面的研究和报道相对较少,尤其是氘代醋酸甲地孕酮,还未见任何专利和文献有过报道。
中国专利申请CN106866767A和CN107501375A虽然分别报道了一种醋酸甲地孕酮的制备方法,但是这两种制备方法无法引入同位素氘,无法获得氘标记的醋酸甲地孕酮。
因此,本领域迫切需要提供一种步骤简单、反应条件温和、成本低、适于大规模生产的氘标记的醋酸甲地孕酮合成方法。
发明内容
本发明的目的是提供一种氘代(氘标记)的醋酸甲地孕酮的合成方法。所述方法所得目标化合物纯度和稳定同位素丰度均能达到98%以上,满足作为定量检测醋酸甲地孕酮的标准试剂的要求,以及在医学研究领域的应用要求;同时步骤简单、反应条件温和、成本低,适于精细化工生产。
本发明为解决上述技术问题而采用的技术方案是提供一种氘标记的醋酸甲地孕酮的合成方法,所述合成方法以17α-羟基孕酮-17-醋酸酯Ⅰ为原料经脱氢反应得到二烯化合物Ⅱ;以二烯化合物Ⅱ为原料经环氧反应得到环氧化合物Ⅲ;以环氧化合物Ⅲ为原料经溴代反应得到溴代化合物Ⅳ;以溴代化合物Ⅳ为原料、氘代甲基硼酸作为氘源,经过Suzuki偶联反应制得氘代醋酸甲地孕酮(化合物Ⅴ)。所得目标化合物纯度和稳定同位素丰度均能达到98%以上。
为解决上述技术问题,本发明采用的技术方案是:一种氘标记醋酸甲地孕酮的合成方法,其特征在于,该方法包括以下步骤:
步骤一、以17α-羟基孕酮-17-醋酸酯Ⅰ为原料,在四氯苯醌、乙酸的氧化作用下脱氢得到二烯化合物Ⅱ;
步骤二、以步骤一所述二烯化合物Ⅱ为原料,在间氯过氧苯甲酸的作用下经环氧反应得到环氧化合物Ⅲ;
步骤三、以步骤二所述环氧化合物Ⅲ为原料,在氢溴酸的作用下经溴代反应得到溴代化合物Ⅳ;
步骤四、以步骤三所述溴代化合物Ⅳ为原料,与氘代甲基硼酸在钯的催化下经Suzuki偶联反应得到氘代醋酸甲地孕酮Ⅴ;
在一种或多种优选的实施方式中,步骤一所述脱氢反应包括:将17α-羟基孕酮-17-醋酸酯Ⅰ、四氯苯醌、乙酸置于容器(如反应瓶)中,开启搅拌,加热至140±10℃(优选地140±5℃或140±3℃或140±2℃)反应6~8h。
在一种或多种优选的实施方式中,所述脱氢反应结束后,还包括分离纯化产物的步骤;更优选地,所述分离纯化产物的步骤包括:静置冷却,通过减压旋蒸法和氢氧化钠溶液中和法除溶剂(减压旋蒸除去大部分溶剂,剩余部分用氢氧化钠溶液中和),过滤得到滤饼,洗涤滤饼除去杂质(优选地用乙酸乙酯洗涤滤饼除去杂质),干燥后得到二烯化合物Ⅱ。
在一种或多种优选的实施方式中,所述17α-羟基孕酮-17-醋酸酯Ⅰ、四氯苯醌和乙酸的摩尔比为1:(1~1.5):(45~60),更优选地为1:(1.05~1.2):(50~55)。
在一种或多种优选的实施方式中,所述脱氢反应采用的溶剂为甲苯。
在一种或多种优选的实施方式中,步骤二所述环氧反应包括:将二烯化合物Ⅱ、间氯过氧苯甲酸(mCPBA)置于容器(如反应瓶)中,开启搅拌,于25~30℃下反应24±6h,优选地24±4h或24±2h或24±1h。
在一种或多种优选的实施方式中,所述环氧反应结束后,还包括分离纯化产物的步骤;更优选地,所述分离纯化产物的步骤包括:滤除固体,将滤液分别使用硫代硫酸钠溶液和碳酸氢钠溶液萃取,干燥后(优选地以无水硫酸钠干燥)减压旋蒸除去溶剂,使用甲醇重结晶,得到白色固体环氧化物Ⅲ。
在一种或多种优选的实施方式中,所述环氧反应采用的溶剂为三氯甲烷;所述二烯化合物Ⅱ和间氯过氧苯甲酸(mCPBA)的摩尔比为1:(1~5),更优选地为1:(2~3),例如为1:2.5。
在一种或多种优选的实施方式中,所述环氧反应采用的溶剂为三氯甲烷或二氯甲烷。
在一种或多种优选的实施方式中,步骤三所述溴代反应包括:将环氧化物Ⅲ、醋酸置于容器(如反应瓶)中,开启搅拌,0~5℃下以1~2滴/秒的速度滴加氢溴酸的醋酸溶液,滴加时间1~3h;滴加完毕后于25~30℃下反应2±0.5h,优选地2±0.3h或2±0.2h。
在一种或多种优选的实施方式中,所述溴代反应结束后,还包括分离纯化产物的步骤;更优选地,所述分离纯化产物的步骤包括:使用氢氧化钠溶液中和,再用二氯甲烷萃取、干燥、减压旋蒸除去溶剂,得到黄色固体,柱层析分离得到溴代化合物Ⅳ。
在一种或多种优选的实施方式中,所述溴代反应中,环氧化物Ⅲ和氢溴酸的摩尔比为1:(2~6),优选地为1:(3~5)。
在一种或多种优选的实施方式中,所述溴代反应中,所述氢溴酸在反应体系中的终浓度为1~50mmol,优选地2~40mmol,更优选地3~30mmol(如4、6、8.8、10、12、15、18、20、25mmol)。
在一种或多种优选的实施方式中,所述氢溴酸为含氢溴酸的乙酸溶液,滴加液中氢溴酸浓度为33±5wt%,优选地33±3wt%,更优选地33±2wt%。
在一种或多种优选的实施方式中,所述溴代反应中,采用的溶剂为乙酸。
在一种或多种优选的实施方式中,步骤四所述Suzuki偶联反应包括:将反应容器(优选地为史莱克瓶)进行无水无氧处理,依次加入溴代化合物Ⅳ、碳酸钾、四三苯基膦钯和氘代甲基硼酸,在100℃下反应36h。
在一种或多种优选的实施方式中,所述Suzuki偶联反应结束后,还包括分离纯化产物的步骤;更优选地,所述分离纯化产物的步骤包括:静置冷却,加水淬灭,二氯甲烷萃取,无水硫酸钠干燥,柱层析得到氘代醋酸甲地孕酮Ⅴ。
在一种或多种优选的实施方式中,所述Suzuki偶联反应中,溴代化合物Ⅳ、氘代甲基硼酸和催化剂钯的摩尔比为1:(1~6):(0.01~0.1),优选地为1:(2~4):(0.02~0.05)。
在一种或多种优选的实施方式中,所述Suzuki偶联反应采用的溶剂为干燥无水的1,4-二氧六环。
在本发明的另一方面,提供前面任一所述的方法的应用,用于制备稳定性氘标记的氘代醋酸甲地孕酮,所述氘代醋酸甲地孕酮的色谱纯度高于98%,同位素丰度高于98%。
本发明提供的氘代标记的醋酸甲地孕酮的合成方法,其主要优点在于:
(1)本发明合成方法中使用的甲苯、氯仿,乙酸等溶剂都比较容易回收再利用,减少了化学废液的处理成本。
(2)本发明各步反应条件温和,没有剧烈放热放气现象,反应安全性好,且反应步骤简单高效。
(3)在步骤四中采用氘代甲基硼酸作为直接氘源,利用Suzuki偶联反应直接上氘甲基的方法减少了氢氘交换的步骤,有效避免了反应中氘代率降低的问题。
附图说明
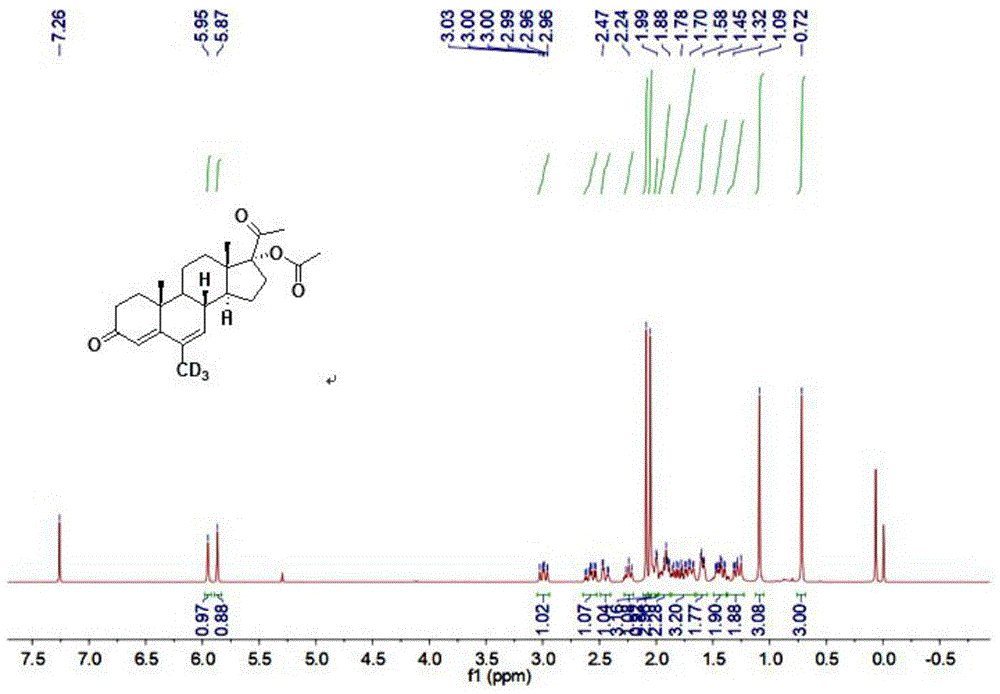
图1为本发明实施例1获得的醋酸甲地孕酮-D3的
图2为本发明实施例1获得的醋酸甲地孕酮-D3的
图3为本发明实施例1获得的醋酸甲地孕酮-D3的液相色谱-质谱图。
具体实施方式
在本发明中,所述的“氘代”与“氘标记”可互换使用。
在本发明中,所有以数值范围或百分比范围形式界定的特征,如数值、数量、含量与浓度仅是为了简洁及方便。据此,数值范围或百分比范围的描述应视为已涵盖且具体公开所有可能的次级范围及范围内的个别数值(包括整数与分数)。
本发明提到的上述特征,或实施例提到的特征可以任意组合。本案说明书所揭示的所有特征可与任何组合物形式并用,只要这些特征的组合不存在矛盾,所有可能的组合都应当认为是本说明书记载的范围。说明书中所揭示的各个特征,可以任何可提供相同、均等或相似目的的替代性特征取代。因此除有特别说明,所揭示的特征仅为均等或相似特征的一般性例子。
为使本发明的目的、技术方案和优点更加清楚,下面将结合本发明的具体实施例及相应的附图对本发明的技术方案进行清楚的描述。显然,所描述的实施例仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
除非另有指明,否则本文所使用的技术及科学上的字词,均为本领域技术人员对于本发明所了解的通常意义,当有冲突情形时,应以本说明书的定义为准。
实施例1
步骤一、以17α-羟基孕酮-17-醋酸酯Ⅰ为原料,在四氯苯醌、乙酸的氧化作用下脱氢得到二烯化合物Ⅱ:
在反应瓶中依次加入17α-羟基孕酮-17-醋酸酯Ⅰ(54mmol)、四氯苯醌(59mmol)、乙酸160mL和甲苯40mL,加热至140℃反应7h,结束后,旋蒸除去大部分溶剂,加入氢氧化钠溶液中和,过滤得到滤饼,用乙酸乙酯洗涤除去杂质,干燥得到二烯化合物Ⅱ,收率90%。
步骤二、以步骤一所述二烯化合物Ⅱ为原料,在间氯过氧苯甲酸(mCPBA)的作用下经环氧反应得到环氧化合物Ⅲ:
在反应瓶中依次加入二烯化合物Ⅱ(14mmol)、mCPBA(28mmol)、100mL氯仿(或者二氯甲烷),于25~30℃下反应24h。反应结束后,滤除固体,将滤液分别使用硫代硫酸钠溶液和碳酸氢钠溶液萃取,无水硫酸钠干燥后减压旋蒸除去溶剂,使用甲醇重结晶,得到白色固体环氧化合物Ⅲ,收率约70%。
步骤三、以步骤二所述环氧化合物Ⅲ为原料,在氢溴酸的作用下经溴代反应得到溴代化合物Ⅳ:
在反应瓶中加入环氧化合物Ⅲ(2.2mmol)和10mL乙酸,0~5℃下以1~2滴/秒的速度滴加氢溴酸(终浓度8.8mmol)的乙酸溶液,滴加时间1h以上,滴加完毕后于25~30℃下反应2h,反应结束后使用氢氧化钠溶液中和,再用二氯甲烷萃取,干燥,旋干,得到黄色固体,柱层析分离,得到溴代化合物Ⅳ,产率为85%。
步骤四、以步骤三所述溴代化合物Ⅳ为原料,与氘代甲基硼酸在钯的催化下经Suzuki偶联反应得到氘代醋酸甲地孕酮Ⅴ:
将史莱克瓶经过三次抽真空换氮后依次加入溴代化合物Ⅳ(0.5mmol)、碳酸钾(1.25mmol)、四三苯基膦钯(0.025mmol)和氘代甲基硼酸(1.5mmol),无水二氧六环5mL。在100℃下反应36h,静置冷却加水猝灭反应,二氯甲烷萃取,无水硫酸钠干燥,最后柱层析得到氘代醋酸甲地孕酮Ⅴ,产率为75%,淡黄色固体,熔点:213.7℃~214.5℃。
制备获得的醋酸甲地孕酮-D3的
实施例2
步骤一、以17α-羟基孕酮-17-醋酸酯Ⅰ为原料,在四氯苯醌、乙酸的氧化作用下脱氢得到二烯化合物Ⅱ:
在反应瓶中依次加入17α-羟基孕酮-17-醋酸酯Ⅰ(20mmol)、四氯苯醌(22mmol)、乙酸80mL和甲苯40mL,加热至140℃反应7h。结束后,旋蒸除去大部分溶剂,加入氢氧化钠溶液中和,过滤得到滤饼,用乙酸乙酯洗涤除去杂质,干燥得到二烯化合物Ⅱ,收率87%。
步骤二、以步骤一所述二烯化合物Ⅱ为原料,在间氯过氧苯甲酸(mCPBA)的作用下经环氧反应得到环氧化合物Ⅲ:
在反应瓶中依次加入二烯化合物Ⅱ(7mmol)、mCPBA(18mmol)、60mL氯仿(或者二氯甲烷),于25~30℃下反应24h。反应结束后,滤除固体,将滤液分别使用硫代硫酸钠溶液和碳酸氢钠溶液萃取,无水硫酸钠干燥后减压旋蒸除去溶剂,使用甲醇重结晶,得到白色固体环氧化合物Ⅲ,收率约75%。
步骤三、以步骤二所述环氧化合物Ⅲ为原料,在氢溴酸的作用下经溴代反应的到溴代化合物Ⅳ:
在反应瓶中加入环氧化合物Ⅲ(5mmol)和10mL乙酸,0~5℃下以1~2滴/秒的速度滴加氢溴酸(终浓度20mmol)的乙酸溶液,滴加时间2h以上,滴加完毕后于25~30℃下反应2h,反应结束后使用氢氧化钠溶液中和,再用二氯甲烷萃取,干燥,旋干,得到黄色固体,柱层析分离,得到溴代化合物Ⅳ,产率为75%。
步骤四、以步骤三所述溴代化合物Ⅳ为原料,与氘代甲基硼酸在钯的催化下经Suzuki偶联反应得到氘代醋酸甲地孕酮Ⅴ:
将史莱克瓶经过三次抽真空换氮后依次加入溴代化合物Ⅳ(1mmol)、碳酸钾(2.5mmol)、四三苯基膦钯(0.025mmol)和氘代甲基硼酸(3mmol),无水二氧六环10mL。在100℃下反应36h,静置冷却加水猝灭反应,二氯甲烷萃取,无水硫酸钠干燥,最后柱层析得到氘代醋酸甲地孕酮Ⅴ,产率为85%,淡黄色固体,熔点:211.7℃~215.5℃。
分析结果显示,色谱纯度和同位素丰度均达到98%以上。
实施例3
步骤一、以17α-羟基孕酮-17-醋酸酯Ⅰ为原料,在四氯苯醌、乙酸的氧化作用下脱氢得到二烯化合物Ⅱ:
在反应瓶中依次加入17α-羟基孕酮-17-醋酸酯Ⅰ(5mmol)、四氯苯醌(7mmol)、乙酸16mL和甲苯40mL,加热至140℃反应7h。结束后,旋蒸除去大部分溶剂,加入氢氧化钠溶液中和,过滤得到滤饼,用乙酸乙酯洗涤除去杂质,干燥得到二烯化合物Ⅱ,收率95%。
步骤二、以步骤一所述二烯化合物Ⅱ为原料,在间氯过氧苯甲酸(mCPBA)的作用下经环氧反应得到环氧化合物Ⅲ:
在反应瓶中依次加入二烯化合物Ⅱ(3mmol)、mCPBA(9mmol)、50mL氯仿(或者二氯甲烷),于25~30℃下反应24h。反应结束后,滤除固体,将滤液分别使用硫代硫酸钠溶液和碳酸氢钠溶液萃取,无水硫酸钠干燥后减压旋蒸除去溶剂,使用甲醇重结晶,得到白色固体环氧化合物Ⅲ,收率约83%。
步骤三、以步骤二所述环氧化合物Ⅲ为原料,在氢溴酸的作用下经溴代反应得到溴代化合物Ⅳ:
在反应瓶中加入环氧化合物Ⅲ(1mmol)和10mL乙酸,0~5℃下以1~2滴/秒的速度滴加氢溴酸(终浓度4mmol)的乙酸溶液,滴加时间1h以上,滴加完毕后于25~30℃下反应2h,反应结束后使用氢氧化钠溶液中和,再用二氯甲烷萃取,干燥,旋干,得到黄色固体,柱层析分离,得到溴代化合物Ⅳ,产率为87%。
步骤四、以步骤三所述溴代化合物Ⅳ为原料,与氘代甲基硼酸在钯的催化下经Suzuki偶联反应得到氘代醋酸甲地孕酮Ⅴ:
将史莱克瓶经过三次抽真空换氮后依次加入溴代化合物Ⅳ(3mmol)、碳酸钾(7.5mmol)、四三苯基膦钯(0.1mmol)和氘代甲基硼酸(9mmol),无水二氧六环50mL。在100℃下反应36h,静置冷却加水猝灭反应,二氯甲烷萃取,无水硫酸钠干燥,最后柱层析得到氘代醋酸甲地孕酮Ⅴ,产率为78%,淡黄色固体,熔点:211.7℃~215.5℃。
分析结果显示,色谱纯度和同位素丰度均达到98%以上。
根据本发明的构思,本领域的技术人员还可以对此作出各种变化和修改,但这些变化和修改均属于本发明权利要求范围以内。
- 一种氮-烷基(氘代烷基)芳杂环和烷基(氘代烷基)芳基醚类化合物的制备方法
- 一种氘代化合物的合成方法
- 一种二氘代伯氨喹的合成方法
- 一种醋酸甲地孕酮合成用催化剂、其制备方法和应用
- 一种由氘气合成氘代乙醇-d6的催化剂、其制备方法及其应用