一种人参益元颗粒HPLC指纹图谱的构建方法及应用
文献发布时间:2024-04-18 20:01:23
技术领域
本发明属于医药领域,具体涉及采用高效液相色谱法建立人参益元颗粒的高效液相指纹图谱用于控制人参益元颗粒的质量。
背景技术
近年来,恶性肿瘤的总体发病率继续呈上升趋势,已成为威胁人类健康的重大疾病。据报道90%以上的化疗药物可能导致骨髓抑制,主要表现为外周血白细胞、血小板、红细胞和血红蛋白出现不同程度的下降,可引起感染、出血和贫血,导致化疗中断,降低患者的生活质量,甚至危及患者生命。目前,西医治疗骨髓抑制的主要药物为集落刺激因子、重组人促血小板生成素及糖皮质激素等,但不良反应多。有研究表明集落刺激因子虽能迅速提高中性粒细胞,但因不能促进造血干细胞增殖而需反复用药,且存在加重肿瘤免疫抑制微环境的风险。恶性肿瘤化疗后的骨髓抑制在中医上属于“血虚”“虚劳”“血证”等范畴,传统中药单味药、经典方剂以益气养血为骨髓抑制的治疗原则,近年来临床研究及报道表明中医药与放化疗药物联用,能起到减毒增效的作用,能使患者按时足量地完成放化疗,改善预后。
人参益元颗粒具有益气补血、扶正培本的功效。用于治疗术后或恶性肿瘤化疗后所致骨髓抑制及免疫功能低下。是由人参,黄芪,白芍、白术、鸡血藤、炙甘草组成。方中药味,人参与黄芪配伍,益元养阴,补中益气,升阳固表,共为君药;白术健脾,助人参、黄芪益气补脾;白芍养血柔肝和营,均为臣药;鸡血藤活血补血,与其他药物共用,奏补而不滞之功,是为佐药;炙甘草为使,益气和中,调和诸药。诸药合用从而达到益气补血、扶正培本的功效。
在本发明完成前,以人参益元颗粒含有的化成分为指标建立指纹图谱的方法未见报道。
发明内容
本发明的目的之一在于,提供一种人参益元颗粒的指纹图谱检测方法及其指纹图谱,采用该方法构建的指纹图谱能够全面地反映人参益元颗粒整体化学成分,为人参益元颗粒的整体质量控制及评价提供了有效手段,旨在解决现有技术中没有关于人参益元颗粒的指纹图谱文献报道的缺陷,有效的控制了人参益元颗粒的质量。
本发明目的之二在于,经过系统的成分指认和药品不同批次共有峰的单味药材归属,较全面的反应人参益元颗粒中各成分的现状,能够为人参益元颗粒质量提供参考依据。
本发明采用如下的技术方案:一种人参益元颗粒的指纹图谱检测方法,采用高效液相色谱法测定其指纹图谱。具体的,所述的人参益元颗粒的指纹图谱检测方法包括以下步骤:
采用80%甲醇对人参益元颗粒进行提取,提取液作为供试品溶液;取原儿茶酸、原儿茶醛、芍药苷、甘草苷、毛蕊花糖苷、染料木素、甘草酸、白术内酯Ⅲ对照品,甲醇溶解,作为对照品溶液;将所述对照品溶液及供试品溶液分别进行高效液相色谱分析,以对照品溶液的色谱峰为基准,得到人参益元颗粒指纹图谱。
优化地,所述人参益元颗粒重量为0.2~2g,80%甲醇的用量为:20~200mL。
优化地,所述提取在超声条件下进行,所述超声的功率为200~300W,频率为30~50kHz;所述提取的时间为20~40min。
优选的,进行所述高效液相色谱分析时,流动相A为乙腈,流动相B为0.02~0.2%的磷酸水溶液;所述流动相体系的流速为0.7~1.2mL/min;采用梯度洗脱方式,梯度洗脱过程中,流动相A、B的比例变化为:0-10min,3%A;10-15min,3-10%A;15-35min,10-15%A;35-65min,15-30%A;65-90min,30-40%A;90-105min,40-80%A。
优选地,进行所述高效液相色谱分析时,以十八烷基硅烷键合硅胶为填充剂,色谱柱规格为:250×4.60mm;柱温为20~40℃;检测波长为210~360nm;进样体积为5~15μL。
优选地,进行所述高效液相色谱分析时,理论塔板数按芍药苷峰计算不低于5000。
优选地,所述人参益元颗粒指纹图谱含有21个共有峰,其中4、5、8、10、12、18、19、21号色谱峰分别被指认为原儿茶酸、原儿茶醛、芍药苷、甘草苷、毛蕊花糖苷、染料木素、甘草酸、白术内酯Ⅲ。
优选地,所述21个共有峰分别归属于白芍、白术、甘草、黄芪、鸡血藤、人参等六味药材;其中1号峰归属于人参、黄芪,2号峰归属于白芍、白术,3号峰归属于白芍、鸡血藤,4号峰归属于鸡血藤,5号峰归属于鸡血藤,6号峰归属于甘草,7号峰归属于白芍,8号峰归属于白芍,9号峰归属于甘草、黄芪,10号峰归属于甘草,11号峰归属于白芍,12号峰归属于黄芪,13号峰归属于人参,14号峰归属于白芍,15号峰归属于甘草、黄芪、鸡血藤,16号峰归属于甘草,17号峰归属于白芍,18号峰归属于鸡血藤,19号峰归属于甘草,20号峰归属于甘草,21号峰归属于白术。
本发明提供了上述技术方案所述人参益元颗粒HPLC指纹图谱的构建方法得到的人参益元颗粒指纹图谱。
本发明提供了上述技术方案所述人参益元颗粒指纹图谱在人参益元颗粒质量控制中的应用。
本发明的有益效果体现在:
1、本发明建立了10批人参益元颗粒的HPLC指纹图谱,相似度均大于0.90,标记了21个共有峰,涵盖了7味中药,可以更加全面的反应人参益元颗粒所含有的化学信息,更好的表征其质量。
2、由于指纹图谱不是为了测定某个成分的精确含量,而是要充分反映化学成分的信息,因此,本发明选择在230nm波长处进行测定,其出峰较多,反映的信息较完全,各个峰吸收值良好,基线平稳。
3、本发明建立的高效液相指纹图谱方法首次实现了对人参益元颗粒全方的质量控制,不是对单一化合物或药材进行鉴别,克服了现有质量控制方法的单一性和片面性问题,能更有效地指导投料、严格规范生产操作,确保临床用药安全、有效。
附图说明
图1混合对照品溶液HPLC色谱图
图2人参益元颗粒共有模式图谱
图3 10个批次人参益元颗粒指纹图谱叠加图
图4样品与各药材色谱图对比归属图
图5 360nm检测波长条件下人参益元颗粒图谱
图6 320nm检测波长条件下人参益元颗粒图谱
图7 280nm检测波长条件下人参益元颗粒图谱
图8 254nm检测波长条件下人参益元颗粒图谱
图9 230nm检测波长条件下人参益元颗粒图谱
图10 210nm检测波长条件下人参益元颗粒图谱
图11 203nm检测波长条件下人参益元颗粒图谱
具体实施方式
本发明中所述人参益元颗粒由人参160g、黄芪480g、白术240g、白芍240g、鸡血藤240g、炙甘草120g制备而成。具体的制备方法为:人参、黄芪两味中药,加10倍量水煎煮二次,第一次3小时,第二次2小时,合并煎液,静置,滤过,滤液浓缩至相对密度为1.30~1.35(60℃)稠膏,备用;其余白术等四味中药,加8倍量水煎煮二次,每次2小时,滤过,滤液浓缩至相对密度为1.10~1.15(60℃)浸膏,加入2倍量乙醇,搅拌,放置12-24小时,滤过,滤液浓缩至相对密度为1.30~1.35(60℃)稠膏,与上述稠膏合并,干燥,粉碎,过筛,加入适量糊精-蔗糖(1:4),混合均匀,用90%乙醇制颗粒,干燥、整粒,制成1000g,即得。
本发明提供了一种人参益元颗粒HPLC指纹图谱的构建方法,包括以下步骤:
a.采用80%甲醇对人参益元颗粒进行提取,将所得人参益元颗粒提取液作为供试品溶液;
b.以原儿茶酸、原儿茶醛、芍药苷、甘草苷、毛蕊花糖苷、染料木素、甘草酸、白术内酯Ⅲ的甲醇溶液作为对照品溶液;
c.将所述对照品溶液及供试品溶液分别进行高效液相色谱分析,以对照品溶液的色谱峰为基准,得到人参益元颗粒指纹图谱。
本发明采用80%甲醇对人参益元颗粒进行提取,将所得人参益元颗粒提取液作为供试品溶液。本发明优选将所述的人参益元颗粒研细后提取。本发明所述人参益元颗粒重量为0.2~2g,80%甲醇的用量为:20~200mL;更优选人参益元颗粒重量为0.5g,80%甲醇的用量为50mL;所述超声的功率为200~300W,频率为30~50kHz,更优选为功率为250W,频率为40kHz;所述提取的时间为20~40min,更优化为30min。
在本发明实施例中,具体是取人参益元颗粒,研细,取0.5g,精密称定,置具塞锥形瓶中,精密加入80%甲醇50mL,称取重量,超声处理(功率40W,频率250kHz)30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得供试品溶液。
本发明的实施例中,具体分别精密称取原儿茶酸、原儿茶醛、芍药苷、甘草苷、毛蕊花糖苷、染料木素、甘草酸铵、白术内酯Ⅲ对照品适量,置10mL量瓶中,以甲醇为溶剂,分别配制成质量浓度分别为0.08034、0.06149、0.9810、0.9672、0.0421、0.05088、0.0436、0.06214mg/mL的对照品储备液溶液;再分别精密吸取对照品储备液0.1、0.1、0.5、0.2、0.1、0.1、0.1、0.1mL置于同一10mL量瓶中,加甲醇溶解并稀释至刻度,摇匀,制成质量浓度分别为0.008034、0.006149、0.04905、0.01934、0.00421、0.005088、0.00436、0.006214mg/mL的混合对照品溶液,过0.45μm的微孔滤膜,取续滤液作为混合对照品溶液。
制备对照品溶液和供试品溶液后,本发明将所述对照品溶液和供试品溶液分别进行高效液相色谱分析,以对照品溶液的色谱峰为基准,得到人参益元颗粒指纹图谱。在本发明中,进行所述高效液相色谱分析时,流动相A为乙腈,流动相B为0.02~0.2%的磷酸水溶液,所述磷酸水溶液进一步优选为0.1%;所述流动相体系的流速为0.7~1.2mL/min,进一步优选为1.0mL/min;采用梯度洗脱方式,梯度洗脱过程中,流动相A、B的比例变化为:0-10min,3%A;10-15min,3-10%A;15-35min,10-15%A;35-65min,15-30%A;65-90min,30-40%A;90-105min,40-80%A。
在本发明中,进行所述高效液相色谱分析时,色谱柱中填充剂优选为十八烷基硅烷键合硅胶,所述色谱柱优选为Alphasil C18柱,规格为:250×4.60mm;柱温为20~40℃,更优选为30℃;检测波长优选为210~360nm,更优选为230nm;进样体积优选为5~15μL,更优选为10μL。
在本发明中,进行所述高效液相色谱分析时,理论塔板数按芍药苷峰计算优选不低于5000,更优选为6000~8000。
将所述对照品溶液和供试品溶液分别进行高效液相色谱分析后,在本发明中,将所述对照品溶液进行高效液相色谱分析后,得到对照品色谱图;将所述供试品溶液进行高效液相色谱分析后,得到样品色谱图;本发明优选将所述样品色谱图导入中药色谱指纹图谱相似度评价系统进行相似度分析,获得人参益元颗粒指纹图谱。在本发明中,所述中药色谱指纹图谱相似度评价系统优选为中药色谱指纹图谱相似度评价系统2012A版;本发明优选利用中位数法进行所述相似度分析,时间窗宽度优选设置为0.1min,共标定21个共有峰。在本发明的实施例中,具体是对10个批次的人参益元颗粒进行高效液相色谱分析,并将所得样品色谱图导入中药色谱指纹图谱相似度评价系统进行相似度分析,各批次人参益元颗粒的相似度均大于0.90。本发明优选将所述样品色谱图与对照品色谱图进行对比,在与各对照品相同保留时间处的色谱峰即得到了指认,结果显示,所述21个共有峰中,其中4、5、8、10、12、18、19、21号色谱峰分别被指认为原儿茶酸、原儿茶醛、芍药苷、甘草苷、毛蕊花糖苷、染料木素、甘草酸、白术内酯Ⅲ。本发明对21个共有峰进行成分归属,所述21个共有峰分别归属于白芍、白术、甘草、黄芪、鸡血藤、人参等六味药材;其中1号峰归属于人参、黄芪,2号峰归属于白芍、白术,3号峰归属于白芍、鸡血藤,4号峰归属于鸡血藤,5号峰归属于鸡血藤,6号峰归属于甘草,7号峰归属于白芍,8号峰归属于白芍,9号峰归属于甘草、黄芪,10号峰归属于甘草,11号峰归属于白芍,12号峰归属于黄芪,13号峰归属于人参,14号峰归属于白芍,15号峰归属于甘草、黄芪、鸡血藤,16号峰归属于甘草,17号峰归属于白芍,18号峰归属于鸡血藤,19号峰归属于甘草,20号峰归属于甘草,21号峰归属于白术。
本发明提供了采用上述技术方案所述人参益元颗粒指纹图谱的构建方法得到的人参益元颗粒指纹图谱。
本发明提供了上述技术方案所述人参益元颗粒指纹图谱在人参益元颗粒质量控制中的应用
为使本发明的技术方案便于理解,以下结合具体试验例对本发明人参益元颗粒的HPLC指纹图谱建立及其指纹图谱作进一步的说明。实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1:检测不同批次的人参益元颗粒的指纹图谱
1、仪器与试药
1.1 Agilent 1220型高效液相色谱仪(美国安捷伦);KQ-250B型超声波清洗器(昆山市超声仪器有限公司);BSA 124S电子天平(赛多利斯);QUINTIX35-1CN(赛多利斯)。
1.2娃哈哈纯净水,乙腈(色谱纯),其他试剂为分析纯。原儿茶酸、原儿茶醛、芍药苷、甘草苷、毛蕊花糖苷、染料木素、甘草酸铵、白术内酯Ⅲ对照品(批号分别为110809-202207、110810-202210、110736-202145、111610-202209、111530-201914、111704-202104、110731-202122、111978-201501,纯度分别为97.5%、99.9%、94.6%、95.2%、95.2%、98.8%、94.4%、99.9%,均购自中国食品药品检定研究院)。人参益元颗粒(批号:220901、220902、220903、221101、221102、221103、230101、230102、230103、230104)。
2、指纹图谱测定
2.1色谱条件:以十八烷基硅烷键合硅胶为填充剂,以乙腈为流动相A,以0.1%磷酸溶液为流动相B,梯度洗脱,流速1.0ml/min,柱温25℃,检测波长230nm,流动相梯度洗脱程序如下:0-10min,3%A;10-15min,3-10%A;15-35min,10-15%A;35-65min,15-30%A;65-90min,30-40%A;90-105min,40-80%A。
2.2供试品溶液的制备:取人参益元颗粒,研细,取0.5g,精密称定,置具塞锥形瓶中,精密加入80%甲醇50mL,称取重量,超声处理(功率40W,频率250kHz)30分钟,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
2.3对照药材及对照提取物的制备:按照处方比例及制备方法,称取对照提取物或对照药材,按照供试品制备方法进行制备。
2.4对照品溶液的制备:分别精密称取原儿茶酸、原儿茶醛、芍药苷、甘草苷、毛蕊花糖苷、染料木素、甘草酸铵、白术内酯Ⅲ对照品适量,精密称定,置10mL量瓶中,以甲醇为溶剂,分别配制成质量浓度分别为0.08034、0.06149、0.9810、0.9672、0.0421、0.05088、0.0436、0.06214mg/mL的对照品储备液溶液;再分别精密吸取对照品储备液0.1、0.1、0.5、0.2、0.1、0.1、0.1、0.1mL置于同一10mL量瓶中,加甲醇溶解并稀释至刻度,摇匀,制成质量浓度分别为0.008034、0.006149、0.04905、0.01934、0.00421、0.005088、0.00436、0.006214mg/mL的混合对照品溶液,过0.45μm的微孔滤膜,取续滤液作为混合对照品溶液;
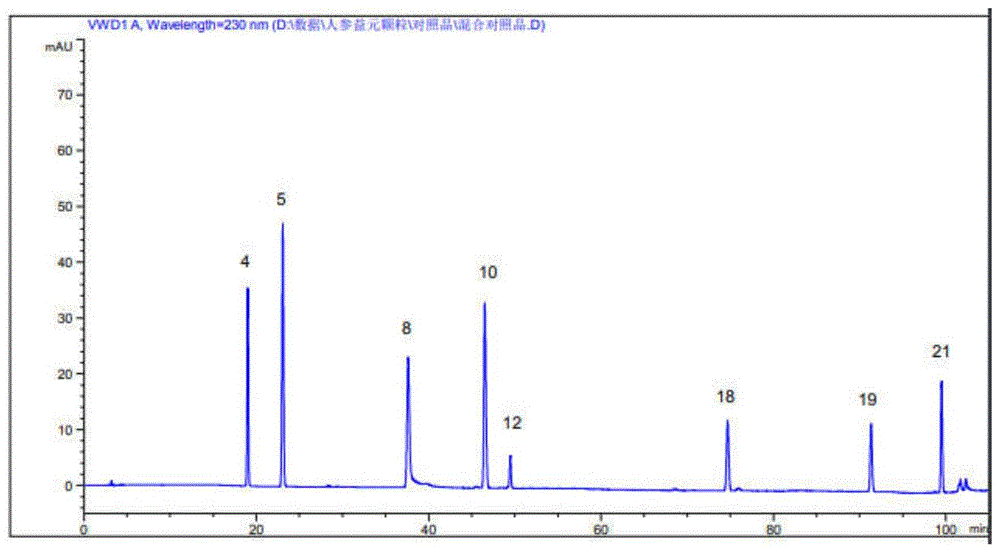
2.4测定:分别精密吸取对照品溶液与供试品溶液各10μL,注入高效液相色谱仪,记录105分钟内的色谱图,分别见图1、图2。
实施例2:10批人参益元颗粒指纹图谱分析
1、指纹图谱相似度分析
取10批次人参益元颗粒,按供试品制备方法制备,色谱条件进样分析,记录人参益元颗粒指纹图谱,采用国家药典委员会发行的《中药色谱指纹图谱相似度评价系统2012A版》对10批次人参益元颗粒组合物指纹图谱进行相似度分析,设定S1为参照图谱,利用中位数法,时间窗宽度设置0.1min,共标定21个共有峰,10批次人参益元颗粒的相似度均大于0.90,相似度评价结果见表1。21个共有峰相对保留时间基本一致,而相对峰面积存在较大差别,见表2。10批次人参益元颗粒指纹图谱见图3(图3中S1-S10分别对应批号220901、220902、220903、221101、221102、221103、230101、230102、230103、230104)。
表1 10批人参益元颗粒样品相似度评价结果
表2标准指纹图谱数据
2、共有峰指认:将对照品所测得的色谱图与样品指纹图谱进行对比,在与各对照品相同保留时间处的色谱峰即得到了指认,对照品色谱图见图1、2(4、5、8、10、12、18、19、21号色谱峰分别被指认为原儿茶酸、原儿茶醛、芍药苷、甘草苷、毛蕊花糖苷、染料木素、甘草酸、白术内酯Ⅲ)。
3、药材归属:按照标准指纹图谱条件进行色谱峰归属,见图4(S1为样品,S2-S7分别为白芍、白术、甘草、黄芪、鸡血藤、人参等六味药材)。21个共有峰分别归属于白芍、白术、甘草、黄芪、鸡血藤、人参等六味药材;其中1号峰归属于人参、黄芪,2号峰归属于白芍、白术,3号峰归属于白芍、鸡血藤,4号峰归属于鸡血藤,5号峰归属于鸡血藤,6号峰归属于甘草,7号峰归属于白芍,8号峰归属于白芍,9号峰归属于甘草、黄芪,10号峰归属于甘草,11号峰归属于白芍,12号峰归属于黄芪,13号峰归属于人参,14号峰归属于白芍,15号峰归属于甘草、黄芪、鸡血藤,16号峰归属于甘草,17号峰归属于白芍,18号峰归属于鸡血藤,19号峰归属于甘草,20号峰归属于甘草,21号峰归属于白术。
实施例3、波长的选择
取220901批样品进行试验,分别考察了360、320、280、250、230、210、203等不同波长下的色谱图的整体效果,其它色谱条件与实施例1相同,见图5、6、7、8、9、10、11。由所测色谱图知,波长为360nm时,色谱峰数量少,所有峰不能全面反映各药材的特征(图5);波长为320nm时,色谱峰数量少,所有峰不能全面反映各药材的特征(图6);波长为280nm时,色谱峰数量少,所有峰不能全面反映各药材的特征(图7);波长为254nm时,色谱峰数量少,所有峰不能全面反映各药材的特征(图8)。波长为230nm时,色谱峰较多,各色谱峰能全面反映各药材特征,色谱图基线较平,峰形较佳,故此为最佳条件(图9)。波长为210时,色谱图基线不平,色谱峰的波动巨大(图10);波长为203nm时,色谱图基线没有处于标准位置(图11)。综合选择,以230nm为检测波长的指纹图谱最能全面反映制剂的成分,而且峰形最好,所以优选230nm为最佳检测波长。
实施例4、方法学考察
取220901批样品进行试验,色谱条件及供试品的制备方法与实施例1相同。
(1)精密度试验
取人参益元颗粒,制备供试品溶液,连续进样6次,记录21个主要色谱峰保留时间和各峰面积。以芍药苷为参照峰,相对峰面积RSD<3.0%(n=6),相对保留时间RSD<1.0%(n=6),证明仪器精密度良好。实验结果见表3。
(2)稳定性试验
取人参益元颗粒,制备供试品溶液,分别于0、2、4、8、12、24h进行测试,记录21个主要色谱峰保留时间和各峰面积。以芍药苷为参照峰,相对峰面积RSD<3.0%(n=6),相对保留时间RSD<1.0%(n=6),证明样品在24h内稳定性良好。实验结果见表3。
(3)重复性试验
取人参益元颗粒,制备供试品溶液6份,进行测试,记录21个主要色谱峰保留时间和各峰面积。以芍药苷为参照峰,相对峰面积RSD<3.0%(n=6),相对保留时间RSD<1.0%(n=6),证明重复性良好。实验结果见表3。
表3方法学考察实验结果
- 一种折叠式防洪装置及折叠式防洪墙
- 一种高精度桥梁主动抬桩预压装置及抬桩方法
- 一种折叠式便携电动滑板车
- 一种便携可折叠式轮胎抬降装置
- 一种便携可折叠式轮胎抬降装置