抗艰难梭状芽胞杆菌的稳定疫苗
文献发布时间:2023-06-19 11:55:48
技术领域
本发明涉及与艰难梭状芽胞杆菌(Clostridium difficile)PS-II细胞表面多糖有关的通式(I)的合成糖及其共轭物。所述合成糖、所述共轭物和包含所述合成糖或所述共轭物的药物组合物是耐水解的、长期稳定的、热稳定的,并且可用于预防和/或治疗与艰难梭状芽胞杆菌[现在被称为艰难梭菌(Clostridioides difficile)]相关的疾病。此外,通式(I)的合成糖在免疫测定中可用作用于检测抗艰难梭状芽胞杆菌的抗体的标记物。
背景技术
艰难梭菌,过去称为艰难梭状芽胞杆菌是革兰氏阳性孢子形成的厌氧菌,其被认为是医院腹泻的最重要的可确定原因。术语艰难梭菌和艰难梭状芽胞杆菌在本文中是同义的,并且都被缩写成C.difficile。它在人的肠道中定植(colonize),从而导致艰难梭状芽胞杆菌感染(CDI)。CDI也已成为医院获得性腹泻的最常见诊断原因,特别是在包括老年人和免疫缺陷患者以及接受抗生素治疗者在内的风险人群中。由于最近十年CDI发病率迅速增加,由C.difficile引起的感染正成为一项重要挑战,这主要归因于超毒力的且现在是主要的027核糖型菌株的出现,从而导致发病率增加、死亡率和高复发率的流行病暴发。治疗费用大大增加,特别是在CDI复发的情况下。因此,非常需要预防由艰难梭状芽胞杆菌引起的感染,并且对危险人群进行疫苗接种是最具成本效益和最有力的手段。然而,尚未开发出针对艰难梭状芽胞杆菌的疫苗。
暴露于细菌细胞表面的碳水化合物通常具有免疫原性,并构成疫苗开发的潜在候选对象。与蛋白质相比,碳水化合物在进化上更稳定,并且当与载体蛋白质共价连接时,寡糖抗原可引发长期的T细胞依赖性保护。
鉴定了由C.difficile细胞表达的三种不同结构的细胞壁多糖,分别称为PS-I、PS-II和PS-III(Expert Rev.Vaccines 2013,12,421)。虽然PS-I糖的表达可能受到更多限制,例如在核糖型027中表达,但是在所有检查的C.difficile核糖型中均发现了PS-II糖,这表明PS-II糖可能是保守的表面抗原。
C.difficile PS-II糖的重复单元由以下组成:
由于(1→6)磷酸二酯键在甘露糖的异头物位置处互连PS-II重复单元的化学稳定性,C.difficile PS-II糖在水中水解,从而使通过常用的热乙酸或水/苯酚从细胞中进行提取变得复杂。裂解O1-C1磷酸二酯键后,除去磷酸单酯基团,从而得到PS-II六糖单元。在存在酸、碱或金属离子的情况下,PS-II糖的磷酸二酯键裂解增加。由于C.difficile PS-II糖在溶液中的不稳定性,当被用作疫苗时,糖或其共轭物必须适当地在液体制剂中缓冲或冻干成固体制剂[其在使用前必须复原(reconstituted)]。然而,疫苗的冻干和冷藏导致增加了生产成本和疫苗递送的复杂性,因为需要一种在运输、储存和处理期间确保最佳温度的有效的冷链系统。C.difficile PS-II糖的不稳定性在本领域中有充分的文献记载。因此,需要液体制剂形式的新的稳定的C.difficile疫苗。
国际专利申请WO 2009/033268 A1公开了从核糖型027、MOH900和MOH718菌株的C.difficile细菌中分离C.difficile的PS-I和PS-II细胞表面糖。Danieli等人(Org.let.2011,13,378–381),Costantino等人(WO 2012/085668 A2),Seeberger(WO2012/119769 A1)和Oberli等人(Chemistry&Biology 2011,18,580)遵循了C.difficile的PS-II细胞表面糖的合成方法。Monteiro(Meth Mol.Biol.2016,397–408)报告了通过热水-苯酚处理从C.difficile生物质中分离水溶性PS-I和PS-II以及水溶性和苯酚可溶性PS-III多糖。
本发明的目的是提供一种明确定义的通式(I)的合成糖,其在液体制剂中是代谢稳定的、抗水解的和货架稳定的(shelf-stable),并且引发了对抗由C.difficile引起的疾病的抗体。所述糖可以与免疫原性载体共轭,以提供可用于预防和/或治疗与C.difficile相关的疾病的共轭物及其药物组合物。此外,通式(I)的合成糖在免疫学测定中可用作用于检测抗C.difficile细菌的抗体的标记物。
本发明的目的通过独立权利要求的教导得以解决。根据本申请的从属权利要求、说明书、附图和实施例,本发明的其他有利特征、方面和细节是显而易见的。
发明内容
定义
如本文所用,术语“接头”涵盖能够任选地通过结合到至少一个互连分子而将糖的还原端单糖与免疫原性载体或固体支持物连接的分子片段。因此,接头本身或与互连分子一起的功能是在还原端单糖与免疫原性载体或固体支持物之间建立、保持和/或桥接特定距离。通过在该糖和免疫原性载体之间保持一定距离,避免了免疫原性载体的结构(例如,载体蛋白的二级结构)对免疫原性糖表位的屏蔽(shielding)。另外,该接头通过减少反应性基团的空间位阻而提供了与糖偶联的更高的效率(Methods in Molecular Medicine2003,87,153-174)。更具体地,接头的一个末端在还原端单糖的异头物中心处与环外氧原子连接,而另一个末端经由氮原子与互连分子连接,或直接与免疫原性载体或固体支持物连接。
本领域已知的糖共轭物(例如糖-载体蛋白共轭物、抗体-药物共轭物)的任何接头均可用于本发明。从大量针对糖载体蛋白共轭物的出版物中,本领域技术人员可以容易地预见用于本文公开的糖和共轭物的合适的接头(见“Antimicrobial glycoconjugatevaccines:an overview of classic and modern approaches for proteinmodification”in Chem Soc Rev 2018,Advance Article,DOI:10.1039/C8CS00495A;以及Acc Chem Res 2017,50,1270-1279),因为所用的接头(即其长度和连接类型)不会显著影响糖共轭物的免疫原性(见PLoS ONE 2017,12(12):e0189100;J.Immun.Meth.1996,191,1-10)。此类合适的接头是无害的(即无毒的)和非免疫原性的(即,在用偶联物免疫后不导致形成非保护性抗体),并且包括但不限于市售的双功能聚乙二醇(Journal of ControlledRelease 2013,172,382-389;J.Immun.Meth.1996,191,1-10),戊二酸衍生物(J.Org.Chem.2005,70(18),7123-7132),己二酸衍生物,方酸衍生物,炔,N-羟基琥珀酰亚胺,例如市售的MFCO-NHS(单氟取代的环辛炔N-羟基琥珀酰亚胺酯),马来酰亚胺(如Acc.Chem.Res.2017,50,1270-1279中公开的),或亲水性亚烷基次膦酸酯和磺酰基(如WO2014080251A1中所述)。
如本文所用,术语“互连分子”是指包含官能团X和官能团Y的双官能分子,其中官能团X能够与接头L上的末端氨基反应并且官能团Y能够与存在于免疫原性载体或固体支持物上的官能团反应。图3显示了市售互连分子的例子,但并不将可根据本发明使用的互连分子限制为本文显示的例子。应当理解,互连分子不形成接头或免疫原性载体或固体支持物的一部分。
如本文所用,术语“佐剂”是指免疫佐剂,即用于疫苗组合物中的材料,其通过增强对疫苗中所含给定抗原的免疫应答而与其没有抗原相关性地来改变或增强所述疫苗的作用。对于本领域技术人员而言,传统上公认的佐剂的例子包括:
-含矿物质的组合物,包括钙盐和铝盐(或其混合物)。钙盐包括磷酸钙。铝盐包括氢氧化物、磷酸盐、硫酸盐等,其中所述盐采取任何合适的形式(例如凝胶、结晶、无定形等)。优选吸附到这些盐上。含矿物质的组合物也可以配制成金属盐的颗粒。也可以使用称为氢氧化铝和磷酸铝的佐剂。本发明可以使用通常用作佐剂的任何“氢氧化物”或“磷酸盐”佐剂。被称为“氢氧化铝”的佐剂通常是氢氧化铝盐,其通常至少部分是结晶的。被称为“磷酸铝”的佐剂通常是羟基磷酸铝,通常还含有少量的硫酸盐(即,羟基磷酸硫酸铝)。它们可以通过沉淀获得,并且在沉淀期间的反应条件和浓度影响磷酸盐取代盐中羟基的程度。在根据本发明的制剂中可以采用氢氧化铝和磷酸铝两者的混合物;
-皂苷,其是在多种植物物种的树皮、叶、茎、根甚至花中发现的固醇糖苷和三萜类糖苷的异源群。来自皂树(Quillaia saponaria),莫利纳树(Molina)的树皮的皂苷已被作为佐剂广泛研究。皂苷也可以购自墨西哥菝葜[Smilax ornata(sarsaprilla)],满天星[Gypsophilla paniculata(brides veil)],以及Saponaria oficianalis(皂根)。皂苷佐剂制剂包括纯化的制剂,例如QS21,以及脂质制剂,例如ISCOM。皂苷组合物已经使用HPLC和RP-HPLC纯化。已经鉴定出使用这些技术的特定纯化的部分(fraction),包括QS 7、QS 17、QS 18、QS21、QH-A、QH-B和QH-C。皂苷制剂还可以包含固醇,例如胆固醇。皂苷和胆固醇的组合可用于形成称为免疫刺激复合物(ISCOM)的独特颗粒。ISCOM通常包括磷脂,例如磷脂酰乙醇胺或磷脂酰胆碱。ISCOM中可以使用任何已知的皂苷。优选地,ISCOM包括QuilA、QHA&QHC中的一个或多个;
-由可生物降解且无毒的材料形成的微粒(即,直径为100nm至150pm,更优选直径为200nm至30pm,或直径为500nm至10pm的颗粒)。此类无毒且可生物降解的材料包括但不限于聚(α-羟基酸)、聚羟基丁酸、聚原酸酯、聚酸酐、聚己内酯;
-CD1d配体,例如α-糖基神经酰胺、含有植物鞘氨醇的α-糖基神经酰胺、OCH、KRN7000[(2S,3S,4R)-1-O-(α-D-半乳吡喃糖基)-2-(N-己糖酰基氨基)-1,3,4-十八烷三醇]、CRONY-101,3"-磺基-半乳糖基-神经酰胺;
-免疫刺激性寡核苷酸,例如包含CpG基序的那些(包含通过磷酸键连接至鸟苷残基的未甲基化胞嘧啶残基的二核苷酸序列),或包含CpI基序的那些(包含连接至肌苷的胞嘧啶的二核苷酸序列),或双链RNA,或包含回文序列的寡核苷酸,或包含多(dG)序列的寡核苷酸。免疫刺激性寡核苷酸可以包括核苷酸修饰/类似物,例如硫代磷酸酯修饰,并且可以是双链的或(除RNA外)是单链的。
-包含与含磷酸盐的无环主链(backbone)连接的脂质的化合物,例如MPLA或TLR4拮抗剂E5564;
-油乳液(例如弗氏佐剂)。
从理论上讲,能够在免疫事件级联(cascade)中促进或放大特定情况并最终导致更明显的免疫应答的每个分子或物质都可以定义为佐剂。
原则上,通过在疫苗制剂中使用佐剂,人们可以:
-指导和优化针对疫苗适合或需要的免疫应答;
-实现疫苗的粘膜递送,即,使疫苗与粘膜表面,如颊或胃或肺上皮及相关淋巴组织接触的给药;
-促进细胞介导的免疫应答;
-增强较弱免疫原(例如高度纯化或重组的抗原)的免疫原性;
-减少提供保护性免疫所需的抗原量或免疫频率;以及
-在免疫应答降低或减弱的个体(例如新生儿、老年人和免疫受损的疫苗接受者)中提高疫苗的功效。
尽管对其作用方式知之甚少,但目前认为佐剂通过以下机制之一增强免疫应答:
-增加抗原的生物学或免疫学半衰期;
-改进抗原向抗原呈递细胞(APC)的递送,以及由APC进行的抗原加工和呈递,例如通过在APC摄取抗原-佐剂复合物后使抗原穿过内体膜进入胞液进行所述改进;
-模拟来自应急或受损细胞的危险诱导信号,这些信号可引发免疫应答;
-诱导免疫调节细胞因子的产生;
-使免疫应答偏向免疫系统的特定亚群;以及-阻断抗原攻击的快速扩散。
糖被本领域技术人员称为TI-2(T细胞非依赖性2)抗原和弱免疫原。因此,为了生产基于糖的疫苗,将所述糖与免疫原性载体共轭以提供共轭物,该共轭物与糖相比具有更高的免疫原性。在本文中,术语“免疫原性载体”定义为与糖共轭以形成与糖本身相比呈现增强的免疫力的共轭物的结构。因此,糖与免疫原性载体(优选蛋白质载体)的共轭实际上刺激了针对所述糖的免疫应答,而没有诱导针对所述免疫原性载体的免疫应答。
因此,本发明涉及通式(I)的糖,
其中,
n是选自1、2、3、4、5、6、7、8、9和10的整数;
T*–表示H–或磷酸基团;
Z表示
L表示接头,并且;
E表示–NH
R′表示–H,–Me,–Et,4-硝基苯基,五氟苯基,或N-琥珀酰亚胺基;
或其非对映异构体或其药学上可接受的盐。
在所有通式(I)、(II)、(III)以及所有子通式中,n优选为1至8的整数,更优选为1至6的整数,并且表示更优选1、2、3、4或5,更优选1、2、3或4,更优选1、2或3,更优选1或2,并且更优选1。
接头L包含优选2至40个碳原子(包括任选的侧链的碳原子),更优选2至30个,更优选2至20个,更优选2至14个,更优选2至12个,并且还更优选2至10个碳原子。
氧原子(即–O–L–NH
还优选的是,接头–L–或最短链完全或部分氟化。接头–L–可以包含3元或4元或5元或6元饱和碳环,或5元部分不饱和的(且不是芳香族的)碳环,或4元或5-元或6元饱和氧杂环,或4元或5元或6元饱和氮杂环,或6元芳族碳环。
接头–L–也可包含酰胺(–NH–CO–,–CO–NH–)和/或尿素(–NH–CO–NH–)残基,优选仅一个酰胺或尿素残基。该接头还可以包含取代基,优选两个取代基,例如R
在接头–L–被氟化的情况下,优选两个以上的取代基–F。
优选地,接头–L–选自:–CH
其中
–L
–L
–L
–L
R
R
o、q、p1和p2彼此独立地是选自1、2、3、4、5、6、7、8、9和10的整数。
更优选地,–L–表示–L
–L
–L
–L
–L
o、q、p1和p2彼此独立地是选自1、2、3、4、5和6的整数。
最优选的是,式(I)的糖具有选自以下的基团–O-L-E:
其中R′表示–H、–Me、–Et、4-硝基苯基,五氟苯基或N-琥珀酰亚胺基;X表示–Br,–Cl,–I,–CO
接头L还可以包含C.difficile PS-II糖的重复单元或其片段:
因此,接头L优选地选自以下结构之一:
因此,优选的还是具有选自以下的基团–O-L-E的式(I)的糖:
或优选该部分的二硫化合物
或优选该部分的二硫化合物
本发明的糖可以是吸湿的并因此可以形成其各种水合物。水分子与糖的摩尔比优选在1至20,更优选在1至10,最优选在5至10的范围内。
本发明的糖带有碱性和/或酸性取代基,并且它们可以与有机或无机酸或碱形成盐。
用于形成这种酸加成盐的合适酸的例子是盐酸,氢溴酸,硫酸,磷酸,乙酸,柠檬酸,草酸,丙二酸,水杨酸,对氨基水杨酸,苹果酸,富马酸,琥珀酸,抗坏血酸,马来酸,磺酸,膦酸,高氯酸,硝酸,甲酸,丙酸,葡萄糖酸,乳酸,酒石酸,羟基马来酸,丙酮酸,苯乙酸,苯甲酸,对氨基苯甲酸,对羟基苯甲酸,甲磺酸,乙磺酸,亚硝酸,羟基乙磺酸,乙磺酸,对甲苯磺酸,萘磺酸,磺胺酸,樟脑磺酸,中国酸,扁桃酸,邻甲基扁桃酸,氢苯磺酸,苦味酸,己二酸,d-邻-甲苯基酒石酸,丙醇二酸(tartronic acid),(邻,间,对)甲苯甲酸,萘胺磺酸,以及本领域技术人员熟知的其他矿物酸或羧酸。通过使游离碱形式与足够量的所需酸接触以常规方式制备盐来制备盐。
合适的无机或有机碱的例子是例如NaOH、KOH、NH4OH、氢氧化四烷基铵、赖氨酸或精氨酸等。可以使用本领域熟知的方法以常规方式制备盐,例如通过用选自上述群组的碱的溶液处理通式(I)的化合物的溶液来制备。
对于碳水化合物化学领域的技术人员显而易见的是,通式(I)的糖不包含–O–O–键和/或经由其异头物或C-1碳相互连接或结合的糖片段。
令人惊讶地,发现通式(I)的糖包含免疫原性保护性表位,并且能够在人和/或动物宿主中诱导针对艰难梭状芽胞杆菌的保护性免疫应答。通式(I)的糖引发与天然艰难梭状芽胞杆菌PS-II细胞表面糖发生交叉反应的抗体,可特异性识别艰难梭状芽胞杆菌细菌并将其调理(opsonize)成被吞噬细胞杀死,从而赋予对抗艰难梭状芽胞杆菌的保护力。
还令人惊讶地发现,通式(I)的糖在酸性水性介质、碱性水性介质以及含有磷酸铝或氢氧化铝的悬浮液(例如常用的佐剂氢氧化铝胶(Alhydrogel))中是稳定的。尽管天然艰难梭状芽胞杆菌PS-II糖在酸性水性介质中,在碱性水性介质中或在铝盐的存在下在一天之内水解,但通式(I)的糖及其共轭物即使在高温下仍可稳定几天。增加的稳定性对于其在抗艰难梭状芽胞杆菌疫苗中的用途是特别有利的。因此,通式(I)的糖及其共轭物特别适用于可在环境温度下储存的对抗艰难梭状芽胞杆菌的货架稳定的液体疫苗制剂。
就纯度和生产容易度而言,本发明的糖克服了与从细菌来源生产的糖及其共轭物有关的所有问题。首先,细胞壁糖的生产需要优化生长条件。其次,需要找到维持构成单糖的结构完整性的解聚条件。最后,需要确定能够分离出限定的长度和结构的纯糖的纯化条件。除了通常的污染物(例如细胞多糖、核酸、脂质和蛋白质)外,还必须排除通过解聚过程获得的不需要的糖。因此,从细菌来源生产具有限定的结构和长度的纯糖是一个繁琐的几乎不可能的过程。
优选的是式(I)或(II)或(III)的合成糖,其中T*–表示磷酸基团(–P(=O)(OH)
T*–表示磷酸基团,即T*–表示–P(=O)(OH)
–P(=O)(O
Z表示
优选地Z表示
L表示接头并且优选为本文公开的接头;
并且其他取代基具有如本文所定义的含义。
优选的是式(I)的合成糖,其中T*–表示氢或磷酸基团(–P(=O)(OH)
T*––表示–H或磷酸基团,即T*–表示–H or–P(=O)(OH)
Z表示
优选地Z表示
L表示接头,并且;
E表示–NH
R′表示–H,–Me,–Et,4-硝基苯基,五氟苯基或N-琥珀酰亚胺基。
优选的是通式(II)的合成糖,
其中n、L、E和T*具有如本文所定义的含义。
因此,其中n、L和E具有本文所定义的含义的通式(II-a)或(II-b)的糖是特别优选的。
通式(III)的合成糖也是优选的,
其中n、L、E和T*具有如本文所定义的含义。
因此,其中n、L和E具有本文所定义的含义的通式(III-a)或(III-b)的糖是特别优选的。
优选地,n表示选自1至10,更优选地1至6,更优选地1至3,甚至更优选地1至2的整数。因此,通式(I),(II),(II-a),(II-b),(III),(III-a)或(III-b)的糖是特别优选的,其中n表示选自1至2的整数。
优选地,接头–L–表示–L
–L
–L
–L
–L
–CH
o、q、p1和p2彼此独立地是选自1、2、3、4、5和6的整数。
因此,通式(I),(II),(II-a),(II-b),(III),(III-a)或(III-b)中任一项的糖是特别优选的,其中,
–L–表示–L
–L
–L
–L
–L
–CH
o、q、p1和p2彼此独立地是选自1、2、3、4、5和6的整数。
通式(I),(II),(II-a),(II-b),(III),(III-a)或(III-b)中任一项的糖也是优选的,其中,
–L–选自:–L
–L
–L
–L
–L
o、q、p1和p2彼此独立地是选自1、2、3、4、5和6的整数;并且n表示1。
甚至更优选的是通式(I),(II),(II-a),(II-b),(III),(III-a)或(III-b)的糖,其中–L–表示–(CH
还优选的是通式(I),(II),(II-a),(II-b),(III),(III-a)或(III-b)的糖,其中–L–表示–(CH
在最优选的实施方案中,–O-L-E选自以下:
其中R′表示–H,–Me,–Et,4-硝基苯基,五氟苯基或N-琥珀酰亚胺基;
X表示–Br,–Cl,–I,–CO
还优选的是通式(I),(II),(II-a),(II-b),(III),(III-a)或(III-b)的糖,其中基团–O-L-E选自以下:
还优选的是通式(I),(II),(II-a),(II-b),(III),(III-a)或(III-b)的糖,其中–L–表示–(CH
优选的是式(II-b)的合成糖,其中n为1且E为氨基。更优选的是式(II-b)的合成糖,其中n为1,E为氨基且接头–L–表示–L
–L
–L
–L
–L
o、q、p1和p2彼此独立地是选自1、2、3、4、5和6的整数。
特别优选的是式(II-b)的合成糖,其中n为1,E为氨基,接头–L–表示–(CH
优选的是式(II-b)的合成糖,其中n为2且E为氨基。更优选的是式(II-b)的合成糖,其中n为2,E为氨基且接头–L–表示–L
–L
–L
–L
–L
o、q、p1和p2彼此独立地是选自1、2、3、4、5和6的整数。
特别优选的是式(II-b)的合成糖,其中n为2,E为氨基,接头–L–表示–(CH
在另一个优选的实施方案中,根据本发明的糖选自以下:
其中Z表示
特别优选的是式(I'a-4)的糖,其中Z表示
特别优选的是式(I'b-4)的糖,其中Z表示
本发明的另一方面涉及合成通式(I)的糖的方法:
其中,
n为1;
T*–表示H–或磷酸基团;
Z表示
L表示接头,并且;
E表示–NH
R′表示–H,–Me,–Et,4-硝基苯基,五氟苯基,或N-琥珀酰亚胺基;
所述方法包括以下步骤:
A1)提供式1*的单糖:
其中P
A2)使式1*的单糖与式2*的化合物反应,得到化合物3*:
其中P
A3)执行化合物3*的保护基P
其中P
A4)使化合物4*与单糖5*反应以得到化合物6*,
其中P
A5)执行化合物6*的保护基P
其中P
A6)使化合物7*与单糖8*反应以得到化合物9*,
其中P
A7)执行化合物9*的保护基P
其中P
A8)使化合物10*与单糖11*反应以得到化合物12*,
其中P
A9)任选地执行化合物12*的保护基P
其中P
A10)将化合物12*或14*的受保护的氨基转化为相应的乙酰氨基以得到化合物15*或16*,
其中P
A11)执行从化合物15*或16*上去除所有剩余的保护基以得到通式(I)的化合物17*或18*,
本发明的另一方面涉及通式(I)的糖17*或18*的合成,其中通过执行步骤A6’直接从化合物7*获得六糖中间体12*。
A6’)使化合物7*与二糖19*反应以得到化合物12*,
其中P
因此,在一个实施方案中,一种合成通式(I)的糖17*或18*的方法包括步骤A1),A2),A3),A4),A5),A6’),A9),A10)和A11)。
本发明的另一方面涉及通式(I)的糖17*或18*的合成,其中通过执行步骤A2’直接从化合物1*获得六糖中间体12*。
A2’)使化合物1与五糖20*反应以得到化合物12*,
其中P
因此,在一个实施方案中,一种合成通式(I)的糖17*或18*的方法包括步骤A1),A2’),A9),A10)和A11)。
化合物1*可以通过步骤A1a),A1b)和A1c)从相应的受保护的甘露糖供体21*获得。
A1a)提供化合物21*,
其中P
将式21*的化合物转化为式22*的醇
其中P
A1b)在磷酸化剂的存在下,使式22*的化合物与醇HO–L–C反应以得到化合物23*;
其中P
步骤A1a)中的醇22*可以根据Brooks等人(Tetrahedron 1995,51,7999)通过在路易斯酸的存在下使化合物21*与烯丙基三甲基硅烷反应(J.Am.Chem Soc.1982,104,4976;Tetrahedron Letters,1985,26,1479),随后在回流的甲苯中用双(苯甲腈)氯化钯(II)异构化为丙烯基C-甘露糖苷,用高碘酸钠和四氧化锇进行Lemieux–Johnson氧化,用乙酰氧基硼氢化钠还原为22*醇(另见Org.Biomol.Chem 2016,14,3913)而制备。
替代地,步骤A1a)中的醇22*可以通过在碘化铜(I)的存在下使化合物21*与(iPrO)Me
在另一个实施方案中,通过与三甲基碘化亚砜和氢化钠反应(J.Org.Chem.2002,67,7439)或通过与炔丙基三甲基硅烷和BF
A1c)执行化合物23*的保护基P
本发明的另一方面涉及合成通式(I)的糖的方法,其中
n是选自2、3、4、5、6、7、8、9或10的整数;
T*–表示H–或磷酸基团;
Z表示
L表示接头,并且;
E表示–NH
R′表示–H,–Me,–Et,4-硝基苯基,五氟苯基,或N-琥珀酰亚胺基;
所述方法包括以下步骤:
B1)提供化合物13*,
其中P
以及重复以下步骤n–1次:
B1.1)在磷酸化剂的存在下与式22*的化合物反应,
B1.2)执行保护基P
B1.3)执行步骤A2)–A8)或步骤A2)–A5)和A6’)或步骤A2’);
B1.4)执行保护基P
或者
B2.1)在磷酸化剂的存在下使化合物13*与下式的化合物反应,
B2.2)执行保护基P
B2.3)任选地重复步骤B2.1和B2.2一至八次以便将相应的三糖(n=3)合成为十糖(n=10);
以提供化合物24*:
其中P
B2)任选地使化合物24*与磷酸化剂反应以得到化合物25*,
其中P
B3)将化合物24*或25*的受保护的氨基转化为相应的乙酰氨基以得到化合物26*或27*,
其中P
B4)执行从化合物26*或27*上去除所有剩余的保护基以得到通式(I)的化合物28*或29*,
其中n表示从2到10的整数并且L和E具有如本文所定义的含义。
本发明的另一方面涉及合成通式(I)的糖的方法,
其中,
n为1;
T*–表示H–或磷酸基团;
Z表示
L表示接头,并且;
E表示–NH
R′表示–H,–Me,–Et,4-硝基苯基,五氟苯基,或N-琥珀酰亚胺基;
所述方法包括以下步骤:
C1)提供式30*的单糖,其可以根据Chem.Eur.J.2015,21,7511-7519或Synlett,2005,7,1147-1151中公开的过程获得:
其中P
C2)使式30*的单糖与式2*的化合物反应以得到化合物31*:
其中P
C3)执行化合物31*的保护基P
其中P
C4)使化合物32*与单糖5*反应以得到化合物33*,
其中P
C5)执行化合物33*的保护基P
其中P
C6)使化合物34*与单糖8*反应以得到化合物35*,
其中P
C7)执行化合物35*的保护基P
其中P
C8)使化合物36*与单糖11*反应以得到化合物37*,
其中P
C9)任选地执行化合物37*的保护基P
其中P
C10)将化合物37*或39*的受保护的氨基转化为相应的乙酰氨基以得到化合物40*或41*,
其中P
C11)执行从化合物40*或41*上去除所有剩余的保护基以得到通式(I)的化合物42*或43*,
本发明的另一方面涉及通式(I)的糖42*或43*的合成,其中通过执行步骤A6’)直接从化合物34*获得六糖中间体37*。因此,在一个实施方案中,一种合成通式(I)的糖42*或43*的方法包括步骤C1),C2),C3),C4),C5),A6’),C9),C10)和C11)。
本发明的另一方面涉及通式(I)的糖42*或43*的合成,其中通过执行步骤A2’直接从化合物30*获得六糖中间体37*。因此,在一个实施方案中,一种合成通式(I)的糖42*或43*的方法包括步骤C1),A2’),C9),C10)和C11)。
因此,另一种用于合成通式(I)的糖的方法包括以下步骤:
C1)提供式30*的单糖:
其中P
C2’)使化合物30*与五糖20*反应以得到化合物37*,
其中P
C9)任选地执行化合物37*的保护基P
其中P
C10)将化合物37*或39*的受保护的氨基转化为相应的乙酰氨基以得到化合物40*或41*,
其中P
C11)执行从化合物40*或41*上去除所有剩余的保护基以得到通式(I)的化合物42*或43*,
其中L和E具有如本文所定义的含义。
化合物30*可以步骤A1a),C1b),C1c)和C1d)从相应的受保护的甘露糖供体21*获得。
C1b)使式22*的化合物转化为相应的卤化物44*
其中P
C1c)在亚磷酸酯(phosphite)的存在下使式44*的化合物与醇HO–L–C反应以得到化合物45*;
其中P
C1d)执行化合物45*的保护基P
在步骤C1b)中醇22*转化为相应的卤化物44*可以根据标准过程实现,该标准过程即通过在PPh
步骤C1c)中使用的亚磷酸酯优选为亚磷酸三烷基酯,例如亚磷酸三乙酯,其与卤化物44*反应生成膦酸酯,接着用路易斯酸(例如溴代三甲基硅烷)然后用水来水解成膦酸(Tetrahedron 1995,51,7999)。在三氯乙腈的存在下,使膦酸与醇HO–L–C反应,以得到化合物45*。
替代地,步骤C1c)中使用的亚磷酸酯可以是亚磷酰胺,例如二烷基或二苄基N,N-二乙基亚磷酰胺或双(二异丙氨基)-苄氧基-膦,其在Arbuzow反应中与化合物44*反应并在二乙胺释放下与醇HO–L–C反应。
本发明的另一方面涉及合成通式(I)的糖的方法,其中,
n是选自2、3、4、5、6、7、8、9或10的整数;
T*–表示H–或磷酸基团;
Z表示
E表示–NH
R′表示–H,–Me,–Et,4-硝基苯基,五氟苯基,或N-琥珀酰亚胺基;
所述方法包括以下步骤:
D1)提供化合物38*,
其中P
D1.1)在亚磷酸酯的存在下与式44*的化合物反应,
D1.2)执行保护基P
D1.3)执行步骤C2)–C8)或步骤C2)–C5)和A6’)或步骤A2’);
D1.4)执行保护基P
或者
D2.1)在偶联剂的存在下使化合物38*与下式化合物反应
D2.2)执行保护基P
D2.3)任选地重复步骤D2.1和D2.2一至八次以便将相应的三糖(n=3)合成为十糖(n=10);
以提供化合物46*:
其中P
D2)任选地使化合物46*与磷酸化剂反应以得到化合物47*,
其中P
D3)将化合物46*或47*的受保护的氨基转化为相应的乙酰氨基以得到化合物48*或49*,
其中P
D4)执行从化合物48*或49*上去除所有剩余的保护基以得到通式(I)的化合物50*或51*,
其中n表示从2到10的整数并且L和E具有如本文所定义的含义。
本发明的另一方面涉及合成通式(I)的糖的方法,其中n为1;
T*–表示H–或磷酸基团;
Z表示
L表示接头,并且;
E表示–NH
R′表示–H,–Me,–Et,4-硝基苯基,五氟苯基,或N-琥珀酰亚胺基;
所述方法包括以下步骤:
E1)提供式52*的单糖:
其中P
E2)使式52*的单糖与式2*的化合物式反应以得到化合物53*:
其中P
E3)执行化合物53*的保护基P
其中P
E4)使化合物54*与单糖5*反应以得到化合物55*,
其中P
E5)执行化合物55*的保护基P
其中P
E6)使化合物56*与二糖19*反应以得到化合物57*,
其中P
E7)将化合物57*的受保护的氨基转化为相应的乙酰氨基以得到化合物58*,
其中P
E8)执行化合物58*的保护基P
其中P
E9)任选地执行化合物15*的保护基P
其中P
E10)执行从化合物15*或16*上去除所有剩余的保护基以得到通式(I)的化合物17*或18*,
本发明的另一方面涉及合成通式(I)的糖的方法,其中,
n是选自2、3、4、5、6、7、8、9或10的整数;
T*–表示H–或磷酸基团;
Z表示
L表示接头,并且;
E表示–NH
R′表示–H,–Me,–Et,4-硝基苯基,五氟苯基,或N-琥珀酰亚胺基;
所述方法包括以下步骤:
F1)提供化合物60*,
其中P
F2.1)在磷酸化剂的存在下使化合物60*与下式的化合物反应
F2.2)执行保护基P
F3)任选地重复步骤F2.1和F2.2 n-2次以便将相应的三聚体(n=3)合成为十聚体(n=10);以提供化合物26*:
其中P
F4)任选地使化合物26*与磷酸化剂反应以得到化合物27*,
其中P
F5)执行从化合物26*或27*上去除所有剩余的保护基以得到通式(I)的化合物28*或29*,
其中n表示从2到10的整数并且L和E具有如本文所定义的含义。
本发明的另一方面涉及合成通式(I)的糖的方法,其中,n是选自1、2、3、4、5、6、7、8、9、10的整数;
T*–表示H–或磷酸基团;
Z表示
L表示接头;并且
E表示–NH
R′表示–H,–Me,–Et,4-硝基苯基,五氟苯基,或N-琥珀酰亚胺基;
所述方法包括以下步骤:
G1)提供式52*的单糖:
其中P
G2)使式52*的单糖与式2*的化合物反应以得到化合物3*:
其中P
G3)执行化合物53*的保护基P
其中P
G4)使化合物54*与单糖5*反应以得到化合物55*,
其中P
G5)执行化合物55*的保护基P
其中P
G6)使化合物56*与二糖19*反应以得到化合物57*,
其中P
G7)将化合物57*的受保护的氨基转化为相应的乙酰氨基以得到化合物58*,
其中P
G8)执行化合物58*的保护基P
其中P
G9)重复步骤G9.1和G9.2 n-1次以便将相应的二聚体(n=3)合成为十聚体(n=10);
G9.1)执行保护基P
G9.2)在磷酸化剂的存在下使步骤G9.1)的产物与下式的化合物反应
,以提供化合物61*,
其中P
G10)任选地执行化合物61*或化合物15*的保护基P
其中P
G11)执行从化合物26*或27*上去除所有剩余的保护基以得到通式(I)的化合物28*或29*,
其中n表示1至10的整数并且L和E具有如本文所定义的含义。
E
N
P
优选的是,可以在分子中存在的其他保护基稳定的条件下除去保护基P
氨基保护基优选在用于除去分子中存在的羟基保护基的条件下是稳定的。
优选除保护基P
更优选地,P
保护基可以区分为永久保护基和临时保护基。永久保护基是在整个合成过程中稳定并且可以在合成后期有效除去的保护基。在这种情况下,永久保护基包括P
临时保护基通常是正交保护基,其可以在合成的不同水平上选择性地除去以释放羟基,以随后引入不同的取代基,包括单糖、分子上存在的其他保护基或其他残基。在这种情况下,临时保护基包括P
临时保护基P
保护基的巧妙选择允许适当得到通式(I),(II),(II-a),(II-b),(III),(III-a)或(III-b),其用末端基团官能化以随后与免疫原性载体或固体支持物进行共轭。此外,离去基团的选择影响步骤A1a),A2),A2’),A4),A6),A6’),A8),B1.3),C2),C4),C6),C8),D1.3),E2),E4)和E6)中糖基化反应的立体化学结果。
构件(Building block)2*,5*,8*,11*,19*,20*和21*是糖基化剂。如本文所用,术语糖基化剂是指在异头位置处用离去基团官能化的单糖,其在用合适的活化剂活化后提供能够与亲核体(例如羟基)反应的氧碳鎓中间体。因此,糖基化剂2*,5*,8*,11*,19*,20*和21*在异头位置处具有离去基团LG
优选地,离去基团LG
优选地,离去基团LG
其中硫醚也可以被取代。
如上所述,氧碳鎓中间体的提供依赖于用合适或恰当的活化剂活化安装在糖基化剂异头位置处的离去基团。本领域技术人员公知的是,适用于磷酸酯的活化剂(即磷酸酯活化剂)和亚氨酸酯(即亚氨酸酯活化剂)是路易斯酸,例如甲硅烷基三氟甲磺酸酯或三氟甲磺酸银,而适用于硫醚的活化剂(即硫醚活化剂)包括但不限于:NIS/TfOH,NIS/TMSOTf,NIS/BF
优选地,LG
优选步骤A1a),A2),A2’),A4),A6),A6’),A8),B1.3),C2),C4),C6),C8),D1.3),E2),E4)和E6)中的糖之间的偶联反应是通过在-10℃至10℃之间的温度下,在非极性溶剂和极性非质子溶剂的混合物中用NIS/TfOH或TMSOTf活化来进行的。甚至更优选的是,通过在约0℃的温度下用NIS/TfOH处理,在非极性溶剂和极性非质子溶剂的混合物中进行。
优选的极性非质子溶剂是四氢呋喃、乙醚和二噁烷。优选的非极性溶剂是甲苯,卤代溶剂,例如氯仿和二氯甲烷。非极性和极性非质子溶剂的优选混合物是:二氯甲烷/四氢呋喃、二氯甲烷/二乙醚、甲苯/二乙醚、甲苯/四氢呋喃。
在步骤A11),B4),C11),D4),E10)和F5)中执行的保护基P
-通过在溶剂混合物中在过氧化氢的存在下用碱处理,第一次裂解对碱不稳定的保护基。优选地,碱是NaOMe或LiOH;以及
-通过在溶剂混合物中在钯催化剂的存在下使化合物经受氢的作用,第二次裂解对氢化敏感的保护基。
步骤A9),B2),C9),D2),E9)和F2.1)中使用的磷酸化剂是能够以其游离形式或以单酯形式将基团P(O)(OH)
在步骤A1b)中使用的磷酸化剂优选为双(二异丙基氨基)-苄氧基膦,苄基N,N-二异丙基膦酰胺酸酯(N,N-diisopropylphosphonamidate)或N,N-二乙基-1,5-二氢-3H-2,3,4-苯并二噁磷-3-胺。在步骤A1b)中使用的优选的活化剂是1H-四唑,4,5-二氰基咪唑,2-苄基硫四唑,5-乙硫基四唑,苯并咪唑三氟甲磺酸酯或咪唑三氟甲磺酸酯。最优选1H-四唑作为活化剂。氧化反应优选在氧化剂(例如过氧化氢或3-氯过苯甲酸)的存在下进行。
根据本发明的另一方面涉及用于制备通式(I),(II),(II-a),(II-b),(III),(III-a)或(III-b)的糖的中间体化合物,其中该中间体化合物具有通式(I2a),(I2b),(I2c),(I2d),(I2e),(I2f),(I4a),(I4b),(I4c),(I4d),(I4e),(I4f),(I4g),(I4h),(I4i),(I4j),(I5a),(I5b),(I5c),(I5d),(I5e),(I5f),(I5g),(I5h),(I5i),(I5j),(I6a),(I6b),(I6c),(I6d),(I6e),(I6f),(I6g),(I6h),(I7a),(I7b),(I7c),(I7d),(I7e),(I7f),(I7g),(I7h),(I7i),(I7j),(I7k),(I7m),(I7n),(I7o)或(I7p)中的任一种:
其中C表示–L–E
更优选的是通式(I),(II),(II-a),(II-b),(III),(III-a)或(III-b)的中间体化合物,其中中间体化合物具有通式(I2a),(I2b),(I2c),(I2d),(I2e),(I2f),(I4a),(I4b),(I4c),(I4d),(I4e),(I4f),(I4g),(I4h),(I4i),(I4j),(I5a),(I5b),(I5c),(I5d),(I5e),(I5f),(I5g),(I5h),(I5i),(I5j),(I6a),(I6b),(I6c),(I6d),(I6e),(I6f),(I6g),(I6h),(I7a),(I7b),(I7c),(I7d),(I7e),(I7f),(I7g),(I7h),(I7i),(I7j),(I7k),(I7m),(I7n),(I7o)或(I7p)中的任一种。
在式(I2a),(I2b),(I2c),(I2d),(I2e),(I2f),(I4a),(I4b),(I4c),(I4d),(I4e),(I4f),(I4g),(I4h),(I4i),(I4j),(I5a),(I5b),(I5c),(I5d),(I5e),(I5f),(I5g),(I5h),(I5i),(I5j),(I6a),(I6b),(I6c),(I6d),(I6e),(I6f),(I6g),(I6h),(I7a),(I7b),(I7c),(I7d),(I7e),(I7f),(I7g),(I7h),(I7i),(I7j),(I7k),(I7m),(I7n),(I7o)或(I7p)中,优选地接头–L–表示–L
–L
–L
–L
–L
–CH
o、q、p1和p2彼此独立地是选自1、2、3、4、5和6的整数。
特别优选的中间体是式(I2a),(I2b),(I2c),(I2d),(I2e),(I2f),(I4a),(I4b),(I4c),(I4d),(I4e),(I4f),(I4g),(I4h),(I4i),(I4j),(I5a),(I5b),(I5c),(I5d),(I5e),(I5f),(I5g),(I5h),(I5i),(I5j),(I6a),(I6b),(I6c),(I6d),(I6e),(I6f),(I6g),(I6h),(I7a),(I7b),(I7c),(I7d),(I7e),(I7f),(I7g),(I7h),(I7i),(I7j),(I7k),(I7m),(I7n),(I7o)或(I7p)的中间体,其中–L–表示–(CH
P
因此,当保护基P
优选地,Np选自–N
本发明的另一方面涉及共轭物,其包含通过–O–L–E基团的末端基团E共价结合或共价连接至免疫原性载体的通式(I)的糖。换句话说,本发明的另一个方面涉及通过–O–L–E基团的末端基团E与免疫原性载体偶联的通式(I),(II),(II-a),(II-b),(III),(III-a)或(III-b)中任一项的糖。包含通过–O–L–E基团的末端基团E共价结合或共价连接至免疫原性载体的通式(I),(II),(II-a),(II-b),(III),(III-a)或(III-b)的共轭物也定义为通过使通式(I),(II),(II-a),(II-b),(III),(III-a)或(III-b)中任一项的糖与免疫原性载体反应而获得的共轭物。令人惊讶地,所述共轭物被证明是用于对抗与艰难梭状芽胞杆菌相关的疾病的免疫的有效的疫苗。
糖被本领域技术人员已知为一般TI-2(T细胞非依赖性-2)抗原和不良的免疫原。TI-2抗原是通过表面暴露的免疫球蛋白受体的交联仅被成熟的B细胞识别的抗原。没有T细胞的帮助,就不会产生免疫记忆,也不会发生从IgM到其他IgG亚类的同种型转换,也不会发生B细胞亲和力成熟。此外,由于与人糖脂和糖蛋白的结构同源性,糖在人体内是不良的免疫原。由于其不良的免疫原性,糖在由B细胞产的抗体和形成记忆细胞两方面(这是生产有效疫苗必不可少的特征)均表现出不佳的能力。
因此,为了生产有效的基于糖的疫苗,通式(I),(II),(II-a),(II-b),(III),(III-a)或(III-b)的糖与免疫原性载体共轭以提供共轭物,与糖相比,所述共轭物呈现增强的免疫原性。因此,在本申请的范围内,还涵盖了包含通过O原子与免疫原性载体共价连接的糖片段的共轭物:
其中n、Z和T*具有本文定义的含义。
所述共轭物包含至少一种通式(I)的合成糖和与至少一种糖(I)共价结合的免疫原性载体。
令人惊讶地发现,用包含与免疫原性载体共价连接的通式(I)的糖的共轭物免疫导致产生对通式(I)的糖的碳水化合物部分具有特异性的高滴度的抗体。所述抗体与天然艰难梭状芽胞杆菌PS-II细胞壁糖发生交叉反应,并呈现调理吞噬和杀菌活性,因此赋予了对抗艰难梭状芽胞杆菌细菌的保护力。
在本文中,术语“免疫原性载体”被定义为一种结构,该结构与糖共轭以形成与糖本身相比呈现增强的免疫原性的共轭物。因此,通式(I),(II),(II-a),(II-b),(III),(III-a)或(III-b)的糖与免疫原性载体的共轭实际上刺激针对通式(I)的糖的免疫应答而不诱导针对所述免疫原性载体的免疫应答。
优选的免疫原性载体是具有免疫调节特性的载体蛋白(CP)或鞘糖脂。对于本领域技术人员而言,载体蛋白(CP)是无毒且无反应原性并且可以足够量和纯度获得的蛋白。载体蛋白选自包括或由以下组成的群组:白喉类毒素,例如CRM
活化的酯包括N-(γ-马来酰亚胺基丁酰氧基)磺基琥珀酰亚胺酯(Sulfo-GMBS),琥珀酰亚胺基(4-碘乙酰基)氨基苯甲酸酯(Sulfo-SIAB),琥珀酰亚胺基-3-(溴乙酰胺基)丙酸酯(SBAP),二琥珀酰亚胺基戊二酰胺酯(DSG),二琥珀酰亚胺基己二酸酯(DSA),2-吡啶基二硫醇-四氧杂十四碳烷-N-羟基琥珀酰亚胺(2-pyridyldithiol-tetraoxatetradecane-N-hydroxysuccinimide,PEG-4-SPDP)(见图2)。
载体蛋白上的半胱氨酸残基可以被转化为相应的脱氢丙氨酸,其可以进一步与合适的互连分子反应以提供在其表面上具有互连分子的官能团X的修饰的载体蛋白。
尤其优选将通式I的糖与无毒的突变白喉毒素CRM
像野生型白喉毒素一样,CRM
因此,在本发明的一个优选实施方案中,载体蛋白在其表面上呈现赖氨酸残基的伯氨基官能团,其能够与互连分子的官能团Y反应以提供在其表面上具有互连分子的所述官能团X的修饰的载体蛋白,其能够与通式(I)的化合物的接头的末端氨基反应。
互连分子的所述官能团X选自包括或由以下组成的群组:马来酰亚胺;α-碘乙酰基;α-溴乙酰基;以及N-羟基-琥珀酰亚胺酯(NHS)、醛、酰亚胺酯、羧酸、烷基磺酸酯、磺酰氯、环氧化物、酸酐、碳酸酯(见图3)。
优选地,通式I的糖被共轭至被马来酰亚胺修饰的无毒的突变白喉毒素CRM
优选的是通式(IV)的共轭物,
其中,
c介于2和18之间;
–E
–W–选自:
a表示选自1、2、3、4、5、6、7、8、9和10的整数,
b表示选自1、2、3和4的整数,
CP是一种载体蛋白;以及
n、L、Z和T*具有如本文所定义的含义。
优选地,E
优选地,CP是CRM
优选地,在通式(IV)中,接头–L–选自:–L
–L
–L
–L
–(CH
–L
以及o、q、p1和p2彼此独立地是选自1、2、3、4、5和6的整数。
而且,通式(IV)的共轭物是优选的,其中–W–表示
尤其优选的是通式(IV)的共轭物,其中
接头–L–选自:–L
–L
–L
–L
–L
o、q、p1和p2彼此独立地是选自1、2、3、4、5和6的整数;
–W–表示
甚至更优选的是通式(IV)的共轭物,其中,
n选自1、2或3;
接头–L–选自:–L
–L
–L
–L
–L
o、q、p1和p2彼此独立地是选自1、2、3、4、5和6的整数;
–W–表示
特别优选的是通式(IV)的共轭物,其中接头–L–表示–(CH
–W–表示
特别优选的是通式(IV)的共轭物,其中n表示1、2或3的整数;
接头–L–表示–(CH
o为选自2、3、4、5和6的整数;
–W–表示
特别优选的是通式(IV)的共轭物,其中n表示1、2或3的整数;
接头–L–表示–(CH
o为选自2、3、4、5和6的整数;
–W–表示
并且Z表示
优选地,c介于2和18之间,更优选地介于5和15之间,甚至更优选地介于8和12之间。还优选地,n表示1。
更优选的是式(IV-1)–(IV-4)中任一项的共轭物:
其中L、E1、W、c、CP和n具有与以上定义相同的含义。
特别优选的是式(IV-2)的共轭物,其中L是–(CH
还优选的是通式(V)的共轭物,
其中,
c介于2和18之间;
–E
–W–选自:
a表示选自1、2、3、4、5、6、7、8、9和10的整数,
b表示选自1、2、3和4的整数;以及
n、L、Z和T*具有如本文所定义的含义。
通式(V)的共轭物是尤其优选的,其中,
接头–L–选自:–L
–L
–L
–L
–L
o、q、p1和p2彼此独立地是选自1、2、3、4、5和6的整数;
–W–表示
甚至更优选的是通式(V)的共轭物,其中,
n选自1、2或3;
接头–L–选自:–L
–L
–L
–L
–L
o、q、p1和p2彼此独立地是选自1、2、3、4、5和6的整数;
–W–表示
特别优选的是通式(V)的共轭物,其中接头–L–表示–(CH
o是选自2、3、4、5和6的整数;
–W–表示
特别优选的是通式(V)的共轭物,其中n表示来自1、2或3的整数;
接头–L–表示–(CH
o是选自2、3、4、5和6的整数;
–W–表示
特别优选的是通式(V)的共轭物,其中n表示来自1、2或3的整数;接头–L–表示–(CH
o是选自2、3、4、5和6的整数;
–W–表示
以及Z表示
特别优选的是通式(V)的共轭物,其中n表示来自1、2或3的整数;
接头–L–表示–(CH
o是选自2、3、4、5和6的整数;
–W–表示
以及T*表示磷酸基团。
还优选的是通式(IV)的共轭物,其中基团–O-L-E选自以下:
更优选的是式(V-1)–(V-4)中任一项的共轭物:
其中L、E
更优选的是式(IV),(IV-1)–(IV-4),(V)和(V-1)–(V-4)中任一项的共轭物,其中n是来自1至3的整数。
更优选的是式(IV),(IV-1)–(IV-4),(V)和(V-1)–(V-4)中任一项的共轭物,其中c选自4至10。
优选地–W–表示
因此,通式(IV),(IV-1)–(IV-4),(V)和(V-1)–(V-4)的共轭物是尤其优选的,其中–W–表示
优选地,接头–L–表示–L
–L
–L
–L
–L
o、q、p1和p2彼此独立地是选自1、2、3、4、5和6的整数。
在最优选的实施方案中,E
还优选的是通式(IV),(IV-1)–(IV-4),(V)和(V-1)–(V-4)的共轭物,其中基团–O-L-E选自以下:
在另一个实施方案中,所述免疫原性载体优选为具有免疫调节特性的鞘糖脂(glycosphingolipid),更优选为(2S,3S,4R)-1-(α-D-半乳吡喃糖基)-2-己糖酰基氨基十八烷基-3,4-二醇。如本文所用,术语具有免疫调节特性的糖鞘脂是指能够刺激免疫系统对靶抗原的应答但其本身不赋予如上定义的免疫力的合适的糖鞘脂。
如本文所用,糖鞘脂是包含与鞘脂α连接的碳水化合物部分的化合物。优选地,碳水化合物部分是六吡喃糖,最优选地是α-D-吡喃半乳糖。对于本领域技术人员而言,鞘脂是一类含有经由酰胺键连接至脂肪酸的C18氨基醇的脂质。C18氨基醇优选被羟基单、双或多取代。特别优选的是,C18氨基醇是植物鞘氨醇。脂肪酸优选为具有碳数范围从16至28,更优选从18至26的饱和烷基链的一元羧酸。具有免疫调节特性的糖鞘脂包括但不限于(2S,3S,4R)-1-(α-D-半乳吡喃糖基)-2-己糖酰基氨基十八烷基-3,4-二醇,其可以刺激自然杀伤T(NK)细胞的自然杀伤(NK)活性和细胞因子的产生,并在体内表现出强大的(Proc.NatlAcad.Sci.USA,1998,95,5690)。
通式I的糖与具有免疫调节特性的鞘糖脂的共轭物具有热稳定的优点。为了适合于共轭,在具有免疫调节特性的鞘糖脂上引入了官能团。所述官能团易于与通式I的糖的接头的末端氨基直接反应以提供通式I的糖的共轭物,或与互连分子的官能团Y直接反应以提供具有免疫调节特性的修饰的糖鞘脂。
优选地,将所述官能团引入具有免疫调节特性的鞘糖脂的碳水化合物部分的C6处。因此,具有免疫调节特性的鞘糖脂用官能团官能化,该官能团易于与糖的末端氨基或与互连分子的官能团Y反应。易于与氨基反应的官能团包括但不限于活化的酯、异氰酸酯基、醛、环氧化物、酰亚胺酯、羧酸、烷基磺酸盐和磺酰氯。易于与互连分子的官能团Y反应以提供具有免疫调节特性从而表现出互连分子的官能团X的修饰的鞘糖脂的官能团包括但不限于胺、醇、硫醇、活化的酯、异氰酸酯基、醛、环氧化物、乙烯基、酰亚胺酯、羧酸、烷基磺酸盐、磺酰氯、乙烯基、炔基和叠氮基。
优选地,在具有免疫调节特性的糖鞘脂的碳水化合物部分的C6处引入的官能团选自包括或包含以下的群组:胺、硫醇、醇、羧酸、乙烯基、马来酰亚胺、α-碘乙酰基、α-溴乙酰基、N-羟基琥珀酰亚胺酯(NHS)、2-吡啶基二硫醇。
互连分子的所述官能团X选自马来酰亚胺、α-碘乙酰基、α-溴乙酰基、N-羟基-琥珀酰亚胺酯(NHS)、醛、羧酸、环氧化物、烷基磺酸盐、磺酰氯、酸酐、碳酸盐。
如本文所用,术语“互连分子”是指包含官能团X和官能团Y的双官能分子,其中官能团X能够与接头–L–上的末端氨基反应并且官能团Y能够与存在于免疫原性载体或固体支持物上的官能团反应。
与包含通过非选择性裂解C.difficile荚膜多糖获得的糖或其共轭物的分离的(而非合成的)混合物的疫苗相比,含有本发明的至少一种共轭物的疫苗引起较少的副作用和/或非保护性免疫应答。此外,与含有非选择性裂解的荚膜多糖的分离混合物的疫苗相比,本发明的疫苗可以根据GMP法规更容易地制造,并且更容易表征,这使得稳定性和纯度控制以及对杂质种类和数量的检测更加容易。
发现包含通式(I),(II),(II-a),(II-b),(III),(III-a)或(III-b)中的任一项的糖的共轭物,特别是通式(IV),(IV-1)–(IV-4),(V)和(V-1)–(V-4)中的任一项的共轭物在人和/或动物宿主体内引发保护性免疫应答,因此可用于预防和/或治疗与艰难梭状芽胞杆菌细菌相关的疾病。因此,包含与免疫原性载体共轭的通式(I)的糖的共轭物可用于预防和/或治疗与艰难梭状芽胞杆菌细菌相关的疾病,所述细菌在其细胞壁糖中含有以下糖片段之一:
-6)-β-D-Glc-(1,3)-β-D-GalNAc-(1,4)-α-D-Glc-(1,4)-[β-D-Glc-(1,3)]-β-D-GalNAc-(1,3)-α-D-Man-(1-;
-3)-α-D-Man-(1,6)-β-D-Glc-(1,3)-β-D-GalNAc-(1,4)-α-D-Glc-(1,4)-[β-D-Glc-(1,3)]-β-D-GalNAc-(1;
-4)-[β-D-Glc-(1,3)]-β-D-GalNAc-(1,3)-α-D-Man-(1,6)-β-D-Glc-(1,3)-β-D-GalNAc-(1,4)-α-D-Glc-(1;
-4)-[β-D-Glc-(1,3)]-β-D-GalNAc-(1,3)-α-D-Man-(1,6)-β-D-Glc-(1,3)-β-D-GalNAc-(1,4)-α-D-Glc-(1;
-3)-β-D-GalNAc-(1,4)-α-D-Glc-(1,4)-[β-D-Glc-(1,3)]-β-D-GalNAc-(1,3)-α-D-Man-(1,6)-β-D-Glc-(1。
优选地,在其细胞壁糖中含有上述糖片段之一的细菌是艰难梭状芽胞杆菌。
在一个优选的实施方案中,包含共轭至免疫原性载体的通式I的糖的共轭物可用于预防和/或治疗与细菌相关的疾病,特别是与在其细胞壁多糖中含有以下糖片段之一的细菌相关的疾病:-6)-β-D-Glc-(1,3)-β-D-GalNAc-(1,4)-α-D-Glc-(1,4)-[β-D-Glc-(1,3)]-β-D-GalNAc-(1,3)-α-D-Man-(1-;-3)-α-D-Man-(1,6)-β-D-Glc-(1,3)-β-D-GalNAc-(1,4)-α-D-Glc-(1,4)-[β-D-Glc-(1,3)]-β-D-GalNAc-(1;-4)-[β-D-Glc-(1,3)]-β-D-GalNAc-(1,3)-α-D-Man-(1,6)-β-D-Glc-(1,3)-β-D-GalNAc-(1,4)-α-D-Glc-(1;-4)-[β-D-Glc-(1,3)]-β-D-GalNAc-(1,3)-α-D-Man-(1,6)-β-D-Glc-(1,3)-β-D-GalNAc-(1,4)-α-D-Glc-(1;-3)-β-D-GalNAc-(1,4)-α-D-Glc-(1,4)-[β-D-Glc-(1,3)]-β-D-GalNAc-(1,3)-α-D-Man-(1,6)-β-D-Glc-(1,并且优选地与艰难梭状芽胞杆菌相关的疾病,其中所述疾病包括腹泻、假膜性结肠炎和麻痹性肠梗阻。
本发明的另一个方面涉及一种药物组合物或疫苗,其包含至少一种共轭物,所述共轭物包含与免疫原性载体共轭的通式(I)的糖和/或通式(I)的糖,以及至少一种药学上可接受的佐剂和/或赋形剂。所述药物组合物可用于在人和/或动物宿主中引起保护性免疫应答。理想地,药物组合物适合用于人类。
在本发明的另一方面,所述药物组合物或疫苗进一步包含艰难梭状芽胞杆菌细菌的至少一种细胞孔糖或细胞壁糖片段和/或其蛋白共轭物,所述细菌选自包含或由以下组成的群组:艰难梭状芽胞杆菌菌株027、MOH718和MOH900。
如本文所用,术语“佐剂”是指免疫佐剂,即用于疫苗组合物中的材料,其通过增强对疫苗中所含给定抗原的免疫应答而与其没有抗原相关性来修饰或增强所述疫苗的作用。对于本领域技术人员而言,免疫佐剂的经典公认例子包括但不限于油乳液(例如弗氏佐剂)、皂苷、铝盐或钙盐(例如明矾)、非离子嵌段聚合物表面活性剂,以及许多其他物质。
药物组合物优选为水性形式,特别是在给药时,但是它们也可以呈现为非水性液体形式或干燥形式,例如明胶胶囊或冻干粉等。
药物组合物可包含一种或多种防腐剂,例如硫柳汞或2-苯氧基乙醇。优选无汞的组合物,并且可以制备无防腐剂的疫苗。
药物组合物可包含生理盐(例如钠盐)以控制张力。氯化钠(NaCl)是典型的,并且可以以介于1和20mg/ml之间的量存在。可能存在的其他盐包括氯化钾、磷酸二氢钾、脱水磷酸二钠、氯化镁、氯化钙等。
药物组合物可以具有介于200mOsm/kg和400mOsm/kg之间的重量克分子渗透浓度(osmolality)。
药物组合物可包括在白水中(例如w.f.i.)的化合物(具有或不具有不溶性金属盐),但是通常将包括一种或多种缓冲液。典型的缓冲液包括:磷酸盐缓冲液;Tris缓冲液;硼酸盐缓冲液;琥珀酸盐缓冲液;组氨酸缓冲液(特别是氢氧化铝佐剂);或柠檬酸盐缓冲液。缓冲盐通常包括在5-20mM的范围内。
药物组合物通常具有介于5.0和9.5之间(例如,介于6.0和8.0之间)的pH。
药物组合物优选是无菌的和无谷蛋白的。
药物组合物适合于施用于动物(尤其是人类)患者,因此包括人类和兽医用途。它们可以用于在患者体内引起免疫应答的方法,包括将该组合物施用于患者的步骤。
本发明的药物组合物可以在受试者暴露于C.difficile之前和/或在受试者暴露于C.difficile细菌之后施用。
在本发明的另一方面,本发明涉及包含共轭至免疫原性载体的至少一种通式(I)的糖和/或至少一种通式(I)的糖的至少一种共轭物用于制造所述药物组合物或所述疫苗的用途,所述药物组合物或所述疫苗用于预防和/或治疗与C.difficile细菌相关的疾病,特别地,与C.difficile细菌相关的疾病选自包含或由以下组成的群组:腹泻、假膜性结肠炎和麻痹性肠梗阻。
优选地,本发明涉及通式(I),(II),(II-a),(II-b),(III),(III-a)或(III-b)中任一项的至少一种糖和/或包含通式(I),(II),(II-a),(II-b),(III),(III-a)或(III-b)中任一项的至少一种糖的至少一种共轭物用于制造所述药物组合物或所述疫苗的用途。
更优选地,本发明涉及糖I'a-1–I'a-11,I'b-1–I'b-11和I'c-1–I'c-11中的至少一种用于制造所述药物组合物或疫苗的用途和/或包含糖I'a-1–I'a-11,I'b-1–I'b-11和I'c-1–I'c-11中的至少一种的至少一种共轭物用于制造所述药物组合物或疫苗的用途。
特别地,本发明涉及通式(IV),(IV-1)–(IV-4),(V)和(V-1)–(V-4)中的任一项的至少一种共轭物用于制造所述药物组合物或所述疫苗的用途。
药物组合物可以制备成单位剂型。在一些实施方案中,单位剂量具有介于0.1-1.0mL之间(例如约0.5mL)的体积。
本发明还提供了包含本发明的药物组合物(例如包含单位剂量)的递送装置(例如注射器、雾化器、喷雾器、吸入器、皮肤贴剂等)。该装置可用于将该组合物施用于脊椎动物受试者。
本发明还提供了包含本发明的药物组合物(例如包含单位剂量)的无菌容器(例如小瓶)。
本发明还提供了本发明的药物组合物的单位剂量。
本发明还提供了包含本发明的药物组合物的气密容器。合适的容器包括例如小瓶。
本发明的药物组合物可以以各种形式制备。例如,可以将组合物制备成注射剂,作为液体溶液或作为悬浮液。也可以制备适于在注射之前溶解或悬浮在液体媒介物中的固体形式(例如冻干的组合物或喷雾冷冻干燥的组合物)。该组合物可以被制备成用于局部给药,例如作为软膏、乳霜或粉末。该组合物可以制备成用于口服给药,例如作为片剂或胶囊剂,作为喷雾剂,或作为糖浆(任选调味的)。该组合物可以被制备成用于肺部给药,例如通过吸入器,使用细粉或喷雾剂。该组合物可以被制备成栓剂。该组合物可以被制备成用于鼻、耳或眼的给药,例如作为喷雾剂或滴剂。用于肌肉内给药的注射剂是典型的。
药物组合物可以包含有效量的佐剂,即当以单剂量或作为系列的一部分施用于个体时有效增强对共同施用的C.difficile PS-II糖抗原的免疫应答的量。
该量可以根据待治疗个体的健康和身体状况、年龄、待治疗个体的分类群(例如非人类灵长类、灵长类等)、个体免疫系统合成抗体的能力、所需的保护程度、疫苗的配方、治疗医生对医疗状况的评估以及其他相关因素而变化。该量将落在可以通过常规试验确定的相对较宽的范围内。
本发明疫苗的配制和给药可以根据本领域中任何已知的方法来实现。
一种根据本发明的共轭物或一种通式(I)的糖的治疗有效剂量是指导致针对疾病的至少部分免疫的化合物的量。这类化合物的毒性和治疗功效可通过细胞培养或实验动物中的标准药物、药理和毒理学程序确定。毒性和治疗效果之间的剂量比是治疗指数。所施加的组合物的实际量将取决于所治疗的受试者、受试者的体重、患病的严重程度、给药方式和处方医师的判断。
本发明的另一方面涉及在人和/或动物宿主中诱导针对C.difficile的免疫应答的方法,所述方法包括将通式(I)的糖和/或其盐和/或其共轭物或其药物组合物施用给所述人和/或动物宿主。根据本发明的在人和/或动物宿主中治疗或预防由C.difficile引起的疾病的方法包括将通式(I)的至少一种糖和/或其盐和/或其共轭物。或其药物组合物施用给所述人和/或动物宿主。
本发明的另一方面涉及通式(I)的糖,其在免疫学测定中用作标记物以检测针对其细胞壁多糖中包含以下糖片段之一的细菌的抗体:
-6)-β-D-Glc-(1,3)-β-D-GalNAc-(1,4)-α-D-Glc-(1,4)-[β-D-Glc-(1,3)]-β-D-GalNAc-(1,3)-α-D-Man-(1-;
-3)-α-D-Man-(1,6)-β-D-Glc-(1,3)-β-D-GalNAc-(1,4)-α-D-Glc-(1,4)-[β-D-Glc-(1,3)]-β-D-GalNAc-(1;
-4)-[β-D-Glc-(1,3)]-β-D-GalNAc-(1,3)-α-D-Man-(1,6)-β-D-Glc-(1,3)-β-D-GalNAc-(1,4)-α-D-Glc-(1;
-4)-α-D-Glc-(1,4)-[β-D-Glc-(1,3)]-β-D-GalNAc-(1,3)-α-D-Man-(1,6)-β-D-Glc-(1,3)-β-D-GalNAc-(1;
-3)-β-D-GalNAc-(1,4)-α-D-Glc-(1,4)-[β-D-Glc-(1,3)]-β-D-GalNAc-(1,3)-α-D-Man-(1,6)-β-D-Glc-(1。
这种测定法包括例如微阵列和ELISA,其可用于检测针对在其细胞壁多糖中包含上述糖片段之一(例如C.difficile)的细菌的抗体。
本发明的糖可以容易地与固体支持物共轭,以提供可用于检测针对C.difficile的抗体的免疫学测定。所述固体载体在其表面上具有官能团,该官能团易于与通式(I)的糖的氨基或与互连分子的官能团Y反应以提供改性的固体载体的,从而在其表面上呈现可以进一步与通式(I)的糖的氨基反应的互连分子的官能团X。在根据本发明的一个实施方案中,固体支持物是微阵列载玻片,其表面上具有易于与互连分子的官能团Y反应以提供修饰的微阵列载玻片的官能团,从而在其表面上呈现互连分子的官能团X。这种微阵列载玻片的例子包括但不限于
在一个优选的实施方案中,固体支持物是微阵列载玻片,其表面上具有易于与通式(I)的糖的氨基,更优选与N-羟基琥珀酰亚胺(NHS)活化的酯反应的官能团。这种微阵列载玻片是例如
附图说明
图1显示了C.difficile PS-II细胞壁糖的重复单元的化学结构。
图2提供了根据本发明的互连分子的官能团X的例子。
图3提供了根据本发明的互连分子的官能团X的例子。
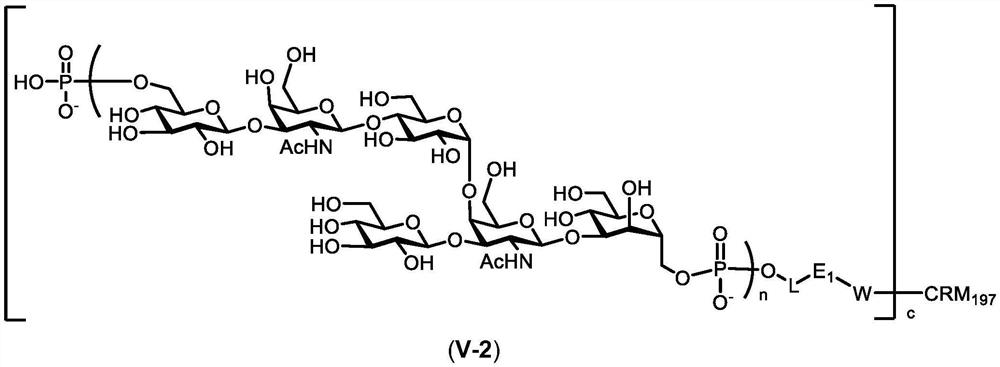
图4显示了作为本申请的优选化合物的通式(V-2)的CRM
图5显示了如何通过NaOH处理裂解化合物33的两条路径。路径I显示了在磷酸酯基团处的裂解,其中磷酸酯基团保留在接头部分,并且形成了化合物LA,5-氨基戊基二氢磷酸酯。路径II显示了在磷酸酯基团上的裂解,其中磷酸酯基团保留在糖部分(化合物33B),并且形成了化合物LB,5-氨基戊烷-1-醇。
图6从下到上显示了以下化合物的HPLC图:化合物33(标准品),化合物33A(对照),在室温下用0.1M氢氧化钠溶液处理一天并纯化后的化合物33,在室温下用0.1M氢氧化钠溶液处理四天后的化合物33,纯化的化合物33B,化合物LB。从图7中可见,化合物33在碱性条件下完全稳定了一天。在室温下用NaOH处理四天后,仍然有50%的化合物33保持完整。
图7从下到上显示了以下化合物的HPLC图:化合物33A(对照),在室温下用0.1M氢氧化钠溶液处理一天并纯化后化合物33,化合物33(标准品),在2℃-8℃含于水中两个月后的化合物33,在2℃-8℃含于NaPi中两个月后的化合物33[其是PBS(磷酸盐缓冲盐水)的同义词],在2℃-8℃含于氢氧化铝胶和PBS中两个月后的化合物33。从图7中可见,化合物33在2℃-8℃在两个月内是完全稳定的。
图8从下到上显示了以下化合物的HPLC图:化合物33A(对照),在室温下用0.1M氢氧化钠溶液处理一天并纯化后的化合物33,化合物33(标准品),在25℃含于水中两个月后的化合物33,在25℃含于NaPi中两个月后的化合物33[其是PBS(磷酸盐缓冲盐水)的同义词],在25℃含于氢氧化铝胶和PBS中两个月后的化合物33。从图8中可见,化合物33在25℃下在两个月内是完全稳定的。
图9从下到上显示了以下化合物的HPLC图:化合物33A(对照),在室温下用0.1M氢氧化钠溶液处理一天并纯化后的化合物33,化合物33(标准品),在37℃含于水中两个月后化合物33,在37℃含于NaPi中两个月后的化合物33[其是PBS(磷酸盐缓冲盐水)的同义词],在37℃含于氢氧化铝胶和PBS中两个月后的化合物33。从图9中可见,化合物33在37℃在两个月内是完全稳定的。
图10从下到上显示了以下化合物的HPLC图:化合物33A(对照),在室温下用0.1M氢氧化钠溶液处理一天并纯化后的化合物33,化合物33(对照),化合物92(标准品),在2-8℃含于水中一周后的化合物92,在2-8℃含于NaPi中一周后的化合物92[其是PBS(磷酸盐缓冲盐水)的同义词],在2-8℃含于氢氧化铝胶和PBS中一周后的化合物92。从图10中可见,化合物92在2-8℃在一周内是完全稳定的。
图11从下到上显示了以下化合物的HPLC图:化合物33A(对照),在室温下用0.1M氢氧化钠溶液处理一天并纯化后的化合物33,化合物33(对照),化合物92(标准品),在25℃含于水中一周后的化合物92,在25℃含于NaPi中一周后的化合物92[其是PBS(磷酸盐缓冲盐水)的同义词],在25℃含于氢氧化铝胶和PBS中一周后的化合物92。从图11中可见,化合物92在25℃在一周内是完全稳定的。
图12从下到上显示了以下化合物的HPLC图:化合物33A(对照),在室温下用0.1M氢氧化钠溶液处理一天并纯化后的化合物33,化合物33(对照),化合物92(标准品),在37℃含于水中一周后的化合物92,在37℃含于NaPi中一周后的化合物92[其是PBS(磷酸盐缓冲盐水)的同义词],在37℃含于氢氧化铝胶和PBS中一周后的化合物92。从图12中可见,化合物92在37℃在一周内是完全稳定的。
图13从下到上显示了以下化合物的HPLC图:化合物33A(对照),在室温下用0.1M氢氧化钠溶液处理一天并纯化后的化合物33,化合物33(对照),化合物54(标准品),在25℃含于水中一周后的化合物54,在2-8℃含于水中一周后的化合物54,在37℃含于水中一周后的化合物54。从图13中可见,化合物54在37℃在一周内是完全稳定的。
图14从下到上显示了以下化合物HPLC图:化合物33A(对照),化合物54(标准品),在25℃含于氢氧化铝胶中一周后的化合物54,在2-8℃含于氢氧化铝胶中一周后的化合物54,在37℃含于氢氧化铝胶中一周后的化合物54。从图14中可见,当用氢氧化铝胶配制时,化合物54大部分被吸附在氢氧化铝上,并且没有形成在氢氧化铝的存在下可通过HPLC检测到的可想到的裂解产物。因此,化合物54在37℃在一周内是稳定的。
图15显示了来自用C.difficile糖33-CRM
图16显示了针对C.difficile菌株630(合并血清)的兔抗血清的ELISA滴度。如图所示,在有或没有氢氧化铝(明矾)佐剂的情况下,每次注射用2.5μg或10μg聚糖抗原对兔(每个研究组4只动物)进行皮下免疫四次(第0、14、28、77天)。具有明矾的PBS用作阴性对照。免疫原是共轭物56。对来自不同时间点(第0、7、21、35、77和84天)的合并血清进行针对涂覆在ELISA板上的福尔马林灭活的C.difficile细菌(菌株630)的总IgG的测试。购自tgcBIOMICS GmbH的涂覆的ELISA板用每孔200μL的商业封闭试剂(Roche,ref.11112589001)封闭2小时。将血清用含于PBS中的1%(w/v)BSA以1:100稀释,并以每孔100μL的体积温育1小时。然后使用HRP共轭山羊抗兔IgG二抗(Sigma-Aldrich,ref.A4914)来检测总IgG,该二抗在含于PBS中的1%(w/v)BSA中30min稀释至1:10,000,并使用TMB底物(Thermo Scientific,ref.34028)显影。在酶标仪中于450nm处测量吸光度,并使用GraphPad Prism软件绘制扣除背景的数据。从图16中可见,用共轭物56接种兔诱导了结合C.difficile细菌菌株630的表面的IgG抗体。此外,添加明矾佐剂导致更高的总体IgG滴度。
图17显示了针对C.difficile菌株630(个体血清)的兔抗血清的ELISA滴度。如图所示,在有或没有氢氧化铝(明矾)佐剂的情况下,每次注射用2.5μg或10μg聚糖抗原对兔(每个研究组4只动物)进行皮下免疫四次(第0、14、28、77天)。具有明矾的PBS用作阴性对照。免疫原是共轭物56。对来自不同时间点(第0、7、35、77和84天)的血清进行针对涂覆在ELISA板上的福尔马林灭活的C.difficile细菌(菌株630)的总IgG的测试。购自tgcBIOMICSGmbH的涂覆的ELISA板用每孔200μL的商业封闭试剂(Roche,ref.11112589001)封闭2小时。将血清用含于PBS中的1%(w/v)BSA以1:300稀释,并以每孔100μL的体积温育1小时。然后使用HRP共轭山羊抗兔IgG二抗(Sigma-Aldrich,ref.A4914)来检测总IgG,该二抗在含于PBS中的1%(w/v)BSA中30min稀释至1:10,000,并使用TMB底物(Thermo Scientific,ref.34028)显影。在酶标仪中于450nm处测量吸光度,并使用GraphPad Prism软件绘制扣除背景的数据。从图17中可见,用共轭物56接种兔诱导了结合C.difficile细菌菌株630的表面的IgG抗体。此外,添加明矾佐剂导致更高的总体IgG滴度。
图18显示了显示了针对C.difficile菌株R20291的兔抗血清的ELISA滴度。如图所示,在有或没有氢氧化铝(明矾)佐剂的情况下,每次注射用2.5μg或10μg聚糖抗原对兔(每个研究组4只动物)进行皮下免疫四次(第0、14、28、77天)。具有明矾的PBS用作阴性对照。免疫原是共轭物56。对来自不同时间点(第21、35、77和84天)的血清进行针对涂覆在ELISA板上的福尔马林灭活的C.difficile细菌(菌株R2029)的总IgG的测试。可商购的涂覆的ELISA板用每孔200μL的商业封闭试剂(Roche,ref.11112589001)封闭2小时。将血清用含于PBS中的1%(w/v)BSA以1:100稀释,并以每孔100μL的体积温育1小时。然后使用HRP共轭山羊抗兔IgG二抗(Sigma-Aldrich,ref.A4914)来检测总IgG,该二抗在含于PBS中的1%(w/v)BSA中30min稀释至1:10,000,并使用TMB底物(Thermo Scientific,ref.34028)显影。在酶标仪中于450nm处测量吸光度,并使用GraphPad Prism软件绘制扣除背景的数据。从图18中可见,用共轭物56接种兔诱导了结合C.difficile细菌菌株R20291的表面的IgG抗体。此外,添加明矾佐剂导致更高的总体IgG滴度。
图19A显示了针对C.difficile菌株VPI10463的兔抗血清(第35天)的ELISA滴度。如图所示,在有或没有氢氧化铝(明矾)佐剂的情况下,每次注射用2.5μg或10μg聚糖抗原对兔(每个研究组4只动物)进行皮下免疫四次(第0、14、28、77天)。具有明矾的PBS用作阴性对照。免疫原是共轭物56。对来自第35天的合并血清进行针对涂覆在ELISA板上的福尔马林灭活的C.difficile细菌(菌株VPI10463)的总IgG的测试。从图19A中可见,用共轭物56接种兔诱导了结合C.difficile细菌菌株VPI10463的表面的IgG抗体。此外,添加明矾佐剂导致更高的总体IgG滴度。
图19B显示了针对C.difficile PS-II多糖的兔抗血清(第35天)的ELISA滴度。如图所示,在有或没有氢氧化铝(明矾)佐剂的情况下,每次注射用2.5μg或10μg聚糖抗原对兔(每个研究组4只动物)进行皮下免疫四次(第0、14、28、77天)。具有明矾的PBS用作阴性对照。免疫原是共轭物56。对来自第35天的合并血清进行针对分离的PS-II多糖的的总IgG的测试。从图19B中可见,用共轭物56接种兔诱导了结合C.difficile细菌菌株VPI10463的表面的IgG抗体。此外,添加明矾佐剂导致更高的总体IgG滴度。
图19C显示了在用分离的C.difficile PS-II多糖预温育或没有进行该预温育的情况下,针对C.difficile菌株630的兔抗血清(第35天)的ELISA滴度。在有氢氧化铝(明矾)佐剂的情况下,每次注射用10μg聚糖抗原对兔(每个研究组4只动物)进行皮下免疫四次(第0、14、28、77天)。将来自第35天的合并血清(在含于PBS中的1%(w/v)BSA中以1:100稀释)在冰上用10或50μg分离的PS-II多糖或用PBS温育30min。然后将该血清在可商购的涂覆的ELISA板(C.difficile菌株630)上温育1小时(100μL/孔),该板已预先用每孔200μL的商业封闭试剂(Roche,ref.11112589001)封闭2小时。然后使用HRP共轭山羊抗兔IgG二抗(Sigma-Aldrich,ref.A4914)来检测总IgG,该二抗在含于PBS中的1%(w/v)BSA中30min稀释至1:10,000,并使用TMB底物(Thermo Scientific,ref.34028)显影。在酶标仪中于450nm处测量吸光度,并使用GraphPad Prism软件绘制扣除背景的数据。从图19C中可见,可以用PS-II多糖以剂量依赖的方式阻断兔抗血清与C.difficile细菌的结合,这表明抗菌抗体应答对PS-II多糖具有特异性。
图20显示了针对C.difficile PS-II六糖54的兔抗血清的ELISA滴度。如图所示,在有或没有氢氧化铝(明矾)佐剂的情况下,每次注射用2.5μg或10μg聚糖抗原对兔(每个研究组4只动物)进行皮下免疫四次(第0、14、28、77天)。具有明矾的PBS用作阴性对照。免疫原是共轭物56。对来自不同时间点(第0、21、35、77和84天)的合并血清进行针对合成的C.difficile PS-II六糖54的总IgG的测试。从图20中可见,用共轭物56接种兔诱导了与合成的免疫原54结合的IgG抗体。此外,添加明矾佐剂导致更高的总体IgG滴度。
图21显示了针对C.difficile菌株630的兔抗血清的ELISA滴度。用共轭物94或共轭物56每次注射以0.5或2μg聚糖抗原的剂量使小鼠(每个研究组7或8只动物)皮下免疫两次(第0、14、28天)。PBS用作阴性对照,并且氢氧化铝(明矾)佐剂用于所有免疫。对来自第21天和第35天的合并血清进行针对涂覆在ELISA板上的福尔马林灭活的C.difficile细菌(菌株630)的总IgG的测试。从图21中可见,用共轭物94或56接种小鼠诱导了结合到C.difficile细菌菌株630的表面的IgG抗体。
图22显示了针对C.difficile菌株R20291的兔抗血清的ELISA滴度。用共轭物94或56每次注射以0.5或2μg聚糖抗原的剂量使小鼠(每个研究组7或8只动物)皮下免疫两次(第0、14、28天)。PBS用作阴性对照,并且氢氧化铝(明矾)佐剂用于所有免疫。对来自第21天和第35天的合并血清进行针对涂覆在ELISA板上的福尔马林灭活的C.difficile细菌(菌株R20291)的总IgG的测试。从图22中可见,用共轭物94或56接种小鼠诱导了结合到C.difficile细菌菌株630的表面的IgG抗体。
图23显示了针对合成的C.difficile PS-II抗原的小鼠抗血清的ELISA滴度。用共轭物94或共轭物56每次注射以0.5或2μg聚糖抗原的剂量使小鼠(每个研究组7或8只动物)皮下免疫两次(第0、14、28天)。PBS用作阴性对照,并且氢氧化铝(明矾)佐剂用于所有免疫。对来自第21天和第35天的合并血清进行针对用于免疫的相应的合成的C.difficile聚糖抗原的总IgG的测试。从图23中可见,用共轭物94或56接种小鼠诱导了结合到合成免疫原的IgG抗体。此外,添加明矾佐剂导致更高的总体IgG滴度。
图24显示了用于免疫实验的两种糖共轭物94和56的SEC色谱图。未共轭的CRM
图25显示了用于用10%聚丙烯酰胺凝胶分离的免疫实验的C.difficile糖共轭物94和56(每孔2.5μg)的SDS-PAGE。未共轭的CRM
包括以下实施例以说明本发明的优选实施方案。本领域技术人员应当理解,以下实施例中公开的技术表示发明人发现的在本发明的实践中发挥良好作用的技术,因此可以认为构成其实践的优选方式。然而,根据本公开,本领域技术人员应当理解,可以在不脱离本发明的精神和范围的情况下对所公开的特定实施方案进行许多改变,并且仍然获得类似或相似的结果。
鉴于该描述,本发明各个方面的进一步修改和替代实施方案对于本领域技术人员将是显而易见的。因此,该描述仅应被解释为说明性的,并且是为了教导本领域技术人员实施本发明的一般方式。应当理解,本文示出和描述的本发明的形式将被视为实施方案的示例。元素和材料可以代替本文中图示和描述的那些,部件和过程可以颠倒,并且本发明的某些特征可以独立地利用,所有这些对于本领域技术人员而言在受益于本发明的该描述之后将是显而易见的。在不脱离如所附权利要求书所述的本发明的精神和范围的情况下,可以对本文所述的元素进行改变。
实施例
除非另有说明,否则使用商品级溶剂。干燥溶剂获自Waters Dry SolventSystem。用于色谱法的溶剂在使用前要进行蒸馏。敏感反应在热干燥的玻璃器皿中和在氩气气氛下进行。在预涂覆有0.25mm厚度硅胶的Kieselgel 60F254玻璃板上进行分析型薄层色谱法(TLC)。通过用香兰素溶液(含于95%乙醇中的6%(w/v)香兰素和10%(v/v)硫酸)或Hanessian氏染色剂(含于水中的5%(w/v)钼酸铵、1%(w/v)硫酸铈(II)和10%(v/v)硫酸)染色。硅胶柱色谱法是在Fluka Kieselgel 60(230-400目)上执行的。
ACN 乙腈
AcOH 醋酸
AIBN 偶氮二异丁腈
氢氧化铝胶 氢氧化铝凝胶佐剂,Al:10mg/mL(Brenntag)
Alloc 烯丙氧羰基
aq. 水性
BH
BBr
Boc 叔丁氧羰基
BnBr 苄基溴
br. 宽
CAS CAS登记号(CAS=化学文摘社)
CHCl
cHex 环己烷
d 双重
dd 双二重
DCM 二氯甲烷
DDQ 2,3-二氯-5,6-二氰基-1,4-苯醌
DEAD 偶氮二羧酸二乙酯
DIPEA N,N-二异丙基-乙胺
DMAP 二甲基氨基吡啶
DME 二甲氧基乙烷
DMF 二甲基甲酰胺
DMSO 二甲基亚砜
DPPA 二苯基磷酰基叠氮化物
EDC·HCl N1-((乙基亚氨基)亚甲基)-N3,N3-二甲基丙烷-1,3-二胺盐酸盐
ES 电喷雾
Et
EtOAc 乙酸乙酯
FCS 胎牛血清
FmocCl 9-芴基甲氧羰基氯化物
GSDMD 消皮素-D
h 小时
HCl 盐酸
HEK293T 胚胎肾成纤维细胞系
H
HOBt.H
hPBMC 人外周血单个核细胞
IC
K
LDH 乳酸脱氢酶
LiAlH
m 多重
MeCN 乙腈
MeOH 甲醇
MeI 甲基碘
MgSO
min 分钟
MS 质谱
Na
NaCNBH
NaHCO
NaH 氢化钠
NaOH 氢氧化钠
NAP 2-萘甲基
NapBr 2-萘甲基溴化物
NaPi缓冲液 磷酸盐缓冲盐水(PBS)
Na
NBS N-溴琥珀酰亚胺
NCS N-氯琥珀酰亚胺
NET 嗜中性粒细胞胞外陷阱
NIS N-碘琥珀酰亚胺
NMR 核磁共振
PBBBr 对溴苄基溴
PBS=NaPi 磷酸盐缓冲盐水
Pd/C 钯碳
Pd(PPh
PMA 佛波醇12-肉豆蔻酸酯13-乙酸酯
PPh
PTFE 聚四氟乙烯
q 四重
RBF 圆底烧瓶
rt 室温
s 单重
sat. 饱和的
sep 七重
t 三重
TBAF 四丁基氟化铵
TFA 三氟乙酸
THF 四氢呋喃
THP1 急性单核细胞白血病癌细胞系
TLC 薄层色谱法
TMSOTf 三甲基甲硅烷基三氟甲磺酸盐
TsOH 对甲苯磺酸(tosic acid)
Wt 重量
合成2
于0℃将NIS(3.0当量)添加到1(根据Chem.Eur.J.2014,20,3578–3583获得)的THF:H
合成3
将Ac
合成4
在室温下,将烯丙基三甲基硅烷(2.0当量)添加到3的干乙腈(20mL/1g)的澄清溶液中,然后逐滴添加TMSOTf(0.5当量)。将烧瓶密封并放置在超声清洗浴中(频率80Hz,100%功率230V,rt),直到通过TLC确认反应完成(40min)。在起始物料完全消耗后,将反应混合物用aq.NaHCO
合成5
将PdCl
合成6
于0℃,将DDQ(1.2当量)添加到5的DCM:H
合成7
将Ac
合成8
于-78℃,将臭氧鼓泡通入7的DCM:MeOH(1:1,170mL/1g)的冷却溶液中,直到持续蓝色。为了除去残留的O
合成9
向8的甲醇(10mL/1g)溶液中添加甲醇钠的MeOH(0.5M,10mL)溶液,并将混合物于rt保持搅拌1h。在8完全消耗后,添加AcOH(1mL),直到反应混合物的pH呈酸性。中和后,将反应混合物浓缩,并将粗残余物通过快速柱色谱法纯化(0-100%EtOAc于环己烷中),得到作为糊状物的期望的化合物9(90%)。针对C
9-化合物10的替代合成
在室温下将炔丙基三甲基硅烷(9.11mL,61.5mmol,2.0当量)添加到3(19.5g,30.8mmol)的干乙腈(390mL)的澄清溶液中,然后逐滴添加TMSOTf(2.8mL,15.4mmol,0.5当量)。将烧瓶密封并放置在超声清洗浴中(频率80Hz,100%功率230V,5-10℃),直到通过TLC(40min)完成反应为止。在起始物料完全消耗后,将反应混合物用aq.NaHCO3淬灭,用EtOAc稀释并用盐水洗涤。分离的有机层在Na
9-化合物11的替代合成
于0℃将DDQ(18.7g,82.0mmol,1.2当量)添加到10(42g,68.5mmol)的DCM:H
9-化合物9的替代合成
于-78℃将臭氧鼓泡通过11(10.6g,22.4mmol)的DCM:MeOH(1:1,1L)的冷却溶液,直到持续蓝色。为了除去残留的O
12的合成
于0℃将氢化钠(2.0当量,含于矿物油中60%)添加到9的THF(20mL/1g)的搅拌溶液中。10min后,添加NapBr(1.05当量),并将混合物于0℃搅拌24h。24h后,将反应混合物用MeOH、水淬灭,并用EtOAc萃取。将合并的有机层用盐水洗涤,在Na
14的合成
于-78℃用新鲜活化的分子筛
15的合成
将FmocCl(2.0当量)和吡啶(3.0当量)添加到14的DCM(10mL/1g)的澄清溶液中,并于rt保持搅拌3.5h。在起始物料完全消耗后,将反应混合物用DCM稀释,并用盐水将其洗涤。有机层在Na
16的合成
于-30℃在
18的合成
于0℃在
19的合成
于-78℃在新鲜活化的分子筛
21的合成
于0℃将氢化钠(2.0当量,含于矿物油中60%)添加到20(根据Tetrahedron:Asymmetry,2000,11,481–492获得)的DMF(10mL/1g)的搅拌溶液中。10min后,添加PBBBr(1.1当量),并将混合物置于rt。于rt搅拌1h后,将反应混合物用NH
22的合成
于0℃将NBS(1.1当量)和TMSOTf(0.1当量)添加到21的DCM:H
23的合成
于0℃将Cs
25的合成
硫代糖苷受体24是根据Danieli,E.;Lay,L.;Proietti,D.;Berti,F.;Costantino,P.;Adamo,R.Org Lett.2011,13,378-381合成的。于-78℃将含于DCM中的TMSOTf(0.1M,0.2当量)添加至含于DCM中的硫代糖苷受体24(1.0当量)和新鲜干燥的
26的合成
于-78℃用新鲜活化的分子筛
27的合成
于0℃将AcCl(2.0当量)和吡啶(3.0当量)添加到26的DCM(10mL/1g)的澄清溶液中,并于rt保持搅拌3.5h。在起始物料完全消耗后,将反应混合物用DCM稀释,并将其用盐水洗涤。有机层在Na
28的合成
于-20℃在
29的合成
向28的EtOAc(2.0mM)的澄清溶液中添加Zn(100当量)和AcOH(100当量),并将反应混合物在室温下搅拌3h。在起始物料完全消耗后,将反应混合物通过硅藻土垫过滤并浓缩。将除去溶剂后获得的残余物溶于EtOAc(2.0mM)中,添加Et
30的合成
将Ac
31的合成
于0℃将DDQ(1.1当量)添加30的DCM:H
32的合成
向31的DCM溶液中添加双(二异丙基氨基)-苄氧基膦(2.0当量)和二异丙基四唑鎓铵(1.5当量),并将该溶液于rt搅拌1.5h。然后,添加5-叠氮基戊醇(8.0当量)和四唑(9.0当量0.45M的CAN溶液),并在室温下保持搅拌2h。2h后,添加叔丁基过氧化物(6.0当量,5.0-6.0M癸烷溶液),并将反应混合物搅拌1h。1h后,将反应混合物用DCM稀释,并用NaHCO
33的合成
将Pd/C(6mg)添加到32(6mg)的EtOAc:MeOH:H
34的合成
将Pd/C(2mg)添加至29的EtOAc:MeOH:H
33与CRM
于rt将抗原33(1.0当量)溶解于2mL小瓶中的DMSO-H
41的合成
将TBDPSCl(1.1当量)和三甲胺(2.8当量)添加到20的CH
42的合成
针对化合物22的合成描述的过程用于化合物42(94%)的合成。针对C
43的合成
于0℃向42的DCM的冷却溶液中添加三氯乙腈(6.0当量)和DBU(0.2当量)。在0℃下3h后,反应完成,并蒸发溶剂。将粗产物通过自动快速硅胶柱色谱法(0-100%EtOAc于环己烷中)纯化,得到作为粘稠液体的期望的产物43(83%)。
44的合成
针对化合物25的合成描述的过程用于化合物44(40%)的合成。针对C
45的合成
针对化合物26的合成描述的过程用于化合物45(60%)的合成。针对C
46的合成
针对化合物27的合成描述的过程用于化合物46(60%)的合成。针对C
47的合成
针对化合物28的合成描述的过程用于化合物47(82%)的合成。针对C
48的合成
针对化合物29的合成描述的过程用于化合物48(50%)的合成。针对C
49的合成
针对化合物30的合成描述的过程用于化合物49(80%)的合成。针对C
50的合成
针对化合物31的合成描述的过程用于化合物50(70%)的合成。针对C
51的合成
针对化合物32的合成描述的过程用于化合物51的合成。
52的合成
于rt将TBAF和AcOH的预混合溶液添加到51的THF澄清溶液中,并将反应混合物于rt搅拌3h。在起始物料完全消耗后,将反应混合物用DCM稀释并在真空下浓缩以获得粗产物。使用含于正己烷中的EtOAc(梯度,0至100%)作为洗脱剂,将粗产物通过自动硅胶柱色谱法纯化。
53的合成
向52的DCM溶液中添加二苄基N,N-二异丙基亚磷酰胺(2.0当量)和二异丙基四唑铵(1.5当量),并将该溶液于rt搅拌1.5h。然后,添加叔丁基过氧化物(6.0当量,5.0-6.0M癸烷溶液),并将反应混合物搅拌1h。1h后,将反应混合物用DCM稀释,并用NaHCO
54的合成
针对化合物33的合成描述的过程用于化合物54的合成。
54与CRM
针对糖共轭物36和37的合成描述的过程用于56和57的合成。
A.4六糖54的替代合成
58的合成
将亚磷酸二苯酯添加到48的吡啶的澄清溶液中,并于室温在氮气下将反应混合物搅拌2h。2h后,于0℃将1M TEAB溶液添加到反应混合物中。5min后,除去冰浴,并于rt继续搅拌另外2h。在起始物料完全消耗后,将反应混合物用DCM稀释,并将有机层依次用1M TEAB溶液洗涤并在减压下浓缩。将粗产物通过自动快速柱色谱法(具有2%Et
59的合成
将H-膦酸酯58(1.0当量)和接头(4.0当量)与吡啶共蒸发,并在真空下干燥30min。之后,将其溶解在py中,并向其中添加PivCl(2.0当量)。将反应混合物于rt保持搅拌2h。2h后,将反应冷却至-40℃,添加新制备的I
60的合成
与0℃向59的DCM和吡啶的溶液中逐滴添加HF溶液(含于吡啶中的70%,0.3mL)。将反应混合物在相同温度下搅拌18h。然后,将反应混合物用DCM稀释,用饱和的NaHCO
54的合成
向60的DCM溶液中添加二苄基二异丙基亚磷酰胺(2.0当量)和二异丙基四唑鎓铵(2.0当量),并将该溶液在室温下搅拌1.5h。然后,在室温下添加叔丁基过氧化物的5.0-6.0M癸烷溶液(6.0当量),并将反应混合物搅拌1h。将反应混合物用DCM稀释,并用NaHCO
62的合成
于0℃将Me
64的合成
针对化合物2的合成描述的过程用于化合物64(85%)的合成。针对C
65的合成
于0℃向64(24.5g,45.3mmol)的无水DCM(360mL)的搅拌溶液中添加无水DMF(1mL,13.6mmol,0.30当量)和(COCl)
66的合成
向糖基氯65(16.2g,28.9mmol,1.15当量)和受体62(13.5g,25.1mmol)的乙腈(200mL)和DCM(80mL)混浊液中添加Ag
67的合成
将FmocCl(16.87g,63.2mmol,2.0当量)和吡啶(7.67mL,95.0mmol,3.0当量)添加到66(33.5g,31.6mmol)的DCM(330mL)澄清溶液中并于rt保持搅拌2h。在起始物料完全消耗后,将反应混合物用DCM稀释,并将其用盐水洗涤。有机层在Na
69的合成
针对化合物2的合成描述的过程用于化合物69(80%)的合成。针对C
70的合成
于0℃向69(18.0g,36.5mmol)的无水DCM(290mL)的搅拌溶液中添加无水DMF(0.85mL,11.0mmol,0.30当量)和(COCl)
71的合成
向糖基氯70(16.5g,32.3mmol,1.15当量)和受体62(15.07g,28.1mmol)的乙腈(200mL)和DCM(80mL)混浊液中添加Ag
72的合成
于0℃下将AcCl(40mL)添加至71(18.87g,18.66mmol)的MeOH(200mL)和DCM(200mL)混浊液中。5min后,移去冰浴,并于rt保持搅拌。在室温下搅拌3h后,将反应混合物用DCM稀释,并用水和aq.NaHCO
73的合成
向72(18.05g,18.6mmol)的乙腈(370mL)悬浮液中添加咪唑(3.56g,52.3mmol,2.8当量)和TBDPSCl(7.2mL,28.0mmol,1.5当量)。5分钟后,反应混合物完全澄清,并置于rt搅拌30分钟。30分钟后,将反应混合物用EtOAc稀释并用盐水洗涤。分离的有机层在Na
74的合成
向73(18.69g,15.47mmol)的DCM(200mL)的澄清溶液中添加Et
76的合成
针对化合物16的合成描述的过程用于化合物76(经2个步骤为60%)的合成。针对C
77的合成
针对化合物18的合成描述的过程用于化合物77(80%)的合成。针对C
78的合成
针对化合物19的合成描述的过程用于化合物78(80%)的合成。针对C
79的合成
针对化合物28的合成描述的过程用于化合物79(79%)的合成。针对C
80的合成
向79的EtOAc(2.0mM)的澄清溶液中添加Zn(100当量)、AcOH(100当量),Ac
81的合成
针对化合物31的合成描述的过程用于化合物81(73%)的合成。针对C
82的合成
针对化合物58的合成描述的过程用于化合物82(87%)的合成。针对C
83的合成
针对化合物59的合成描述的过程用于化合物83(88%)的合成。针对C
84的合成
针对化合物60的合成描述的过程用于化合物84(90%)的合成。针对C
33的合成
针对从化合物32合成33描述的过程用于化合物33(55%)的合成。针对C
85的合成
向84的DCM溶液中添加二苄基N,N-二异丙基亚磷酰胺(2.0当量)和二异丙基四唑鎓铵(1.5当量),并将该溶液于rt搅拌2.5h。然后,添加叔丁基过氧化物(6.0当量,5.0-6.0M癸烷溶液),并将反应混合物搅拌1h。1h后,将反应混合物用DCM稀释,并用NaHCO
54的合成
将Pd/C(20mg)添加到85(20mg)的EtOAc:MeOH:H
86的合成
针对化合物58的合成描述的过程用于化合物86(94%)的合成。针对C
87的合成
将H-膦酸酯86(1.0当量)和苯甲醇(10.0当量)与吡啶共蒸发,并在真空下干燥30min。之后,将其溶解在py中,并向其中添加PivCl(5.0当量)。将反应混合物于rt保持搅拌2h。2h后,将反应冷却至-40℃,添加新制备的I
54的合成
将Pd/C(20mg)添加到87(20mg)的EtOAc:MeOH:H2O:DCM的澄清溶液中。将获得的非均质混合物在氢气氛下于rt搅拌40h。40h后,将反应混合物通过PTFE过滤器过滤,并于旋转蒸发器30℃浴温在真空下浓缩10min,以除去甲醇、EtOAc、DCM和水。将除去溶剂后获得的粗产物溶解于MeOH、水,并于0℃向其中添加LiOH(2N于水中)。于0℃将反应混合物搅拌3h。3h后,将反应混合物用AcOH淬灭,并在减压下除去溶剂,并使用水和乙腈作为溶剂,通过C18反相柱色谱法将所获得的粗残余物纯化,得到盐形式的期望的最终化合物54。然后将三乙胺盐与Dowex树脂交换,得到作为白色固体的具有钠盐的期望的化合物(经3个步骤为70%)。针对C
88的合成
针对化合物32的合成描述的过程用于化合物88的合成,这里唯一的变化是,在第二步骤中,化合物52取代接头用作亲核试剂。
89的合成
针对化合物52的合成描述的过程用于化合物89的合成。
90的合成
针对化合物33的合成描述的过程用于化合物90的合成。
91的合成
针对化合物53的合成描述的过程用于化合物91的合成。
92的合成
针对化合物33的合成描述的过程用于化合物92(60%)的合成。针对C
92与CRM
针对糖共轭物36和37的合成描述的过程用于94和95的合成。
将H-膦酸酯58(1.2当量)和受体60(1.0当量)与吡啶共蒸发,并在真空下干燥30min。之后,将其溶解在py中,并向其中添加PivCl(1.3当量)。将反应混合物于rt保持搅拌3h。3h后,将反应冷却至-40℃,添加新鲜制备的I
97的合成
针对化合物60的合成描述的过程用于化合物97(70%)的合成。针对C
92的合成
针对化合物61的合成描述的过程用于化合物98(89%)的合成。针对化合物54的合成描述的过程用于化合物92(60%)的合成。针对C
99的合成
于0℃将MsCl和吡啶(py)添加到8的DCM澄清溶液中。将反应混合物在室温下搅拌过夜,然后用DCM稀释,用aq.NaHCO
100和101的合成
将碘化钠添加到99的2-丁酮澄清溶液中,并于100℃将反应混合物搅拌过夜。然后,除去溶剂,并将粗残余物溶解在DCM中,用aq.NaHSO
102的合成
将TEA和苯硫酚添加到101的THF澄清溶液中。将反应混合物在室温搅拌24h。起始物料完全耗尽后,将反应混合物用TEA稀释并浓缩,得到粗残余物,并将其通过硅胶色谱法纯化,得到102。
103的合成
将膦酸酯102、接头和三苯膦溶解在THF中,并将溶液冷却至0℃,然后向其中添加DIAD。将混合物在室温下搅拌24h。24h后,将溶液浓缩,并将粗产物通过硅胶色谱法纯化,得到103。
104的合成
将TEA和苯硫酚添加到103的THF澄清溶液中。将反应混合物在室温下搅拌24h。起始物料完全耗尽后,将反应混合物用TEA稀释并浓缩,得到粗残余物,并通过硅胶色谱法将其纯化,得到104。
105的合成
将膦酸酯衍生物104溶解于0.05M NaOMe的MeOH溶液中,并于rt搅拌10min。然后将反应混合物用AcOH淬灭,并在真空下除去溶剂。通过硅胶色谱法纯化所获得的粗残余物,得到105。
106的合成
根据化合物16的合成进行反应。
107的合成
根据化合物18的合成进行反应。
108的合成
根据化合物19的合成进行反应。
109的合成
根据化合物28的合成进行反应。
110的合成
根据化合物29的合成进行反应。
111的合成
根据化合物30的合成进行反应。
112的合成
根据化合物33的合成进行反应。
112与CRM197或BSA的共轭
根据化合物33的共轭进行反应。
116的合成
将H-膦酸酯58和接头与吡啶共蒸发,并在真空下干燥30min。之后,将其溶解在吡啶中,并向其中添加PivCl。将反应混合物于rt保持搅拌2h。2h后,将反应冷却至-40℃,添加新鲜制备的I
117的合成
根据化合物31的合成进行反应。
117与CRM
根据化合物33的共轭进行反应。
121的合成
将H-膦酸酯58和接头与吡啶共蒸发,并在真空下干燥30min。之后,将其溶解在吡啶中,并向其中添加PivCl。将反应混合物于rt保持搅拌2h。2h后,将反应冷却至-40℃,添加新鲜制备的I
122的合成
根据化合物33的合成和TBS脱保护步骤进行反应。
122与CRM
根据化合物33的共轭进行反应。
126的合成
将H-膦酸酯58和接头与吡啶共蒸发,并在真空下干燥30min。之后,将其溶解在吡啶中,并向其中添加PivCl。将反应混合物于rt保持搅拌2h。2h后,将反应冷却至-40℃,添加新鲜制备的I
127的合成
根据化合物33的合成和TBS脱保护步骤进行反应。
127与CRM
根据化合物33的共轭进行反应。
131的合成
将H-膦酸酯58和接头与吡啶共蒸发,并在真空下干燥30min。之后,将其溶解在吡啶中,并向其中添加PivCl。将反应混合物于rt保持搅拌2h。2h后,将反应冷却至-40℃,添加新鲜制备的I
132的合成
根据化合物33的合成和TBS脱保护步骤进行反应。
132与CRM
根据化合物33的共轭进行反应。
136的合成
将H-膦酸酯58和接头与吡啶共蒸发,并在真空下干燥30min。之后,将其溶解在吡啶中,并向其中添加PivCl。将反应混合物于rt保持搅拌2h。2h后,将反应冷却至-40℃,添加新鲜制备的I
137的合成
根据化合物33的合成和TBS脱保护步骤进行反应。
137与CRM
根据化合物33的共轭进行反应。
141的合成
将H-膦酸酯58和接头与吡啶共蒸发,并在真空下干燥30min。之后,将其溶解在吡啶中,并向其中添加PivCl。将反应混合物于rt保持搅拌2h。2h后,将反应冷却至-40℃,添加新鲜制备的I
142的合成
根据化合物33的合成和TBS脱保护步骤进行反应。
142与CRM
根据化合物33的共轭进行反应。
146的合成
将H-膦酸酯58和接头与吡啶共蒸发,并在真空下干燥30min。之后,将其溶解在吡啶中,并向其中添加PivCl。将反应混合物于rt保持搅拌2h。2h后,将反应冷却至-40℃,添加新鲜制备的I
142的合成
根据化合物33的合成和TBS脱保护步骤进行反应。
根据化合物33的共轭进行反应。
151的合成
将H-膦酸酯58和接头与吡啶共蒸发,并在真空下干燥30min。之后,将其溶解在吡啶中,并向其中添加PivCl。将反应混合物于rt保持搅拌2h。2h后,将反应冷却至-40℃,添加新鲜制备的I
152的合成
根据化合物33的合成和TBS脱保护步骤进行反应。
152与CRM
在室温下将SBAP(N-琥珀酰亚胺基-3-(溴乙酰氨基)丙酸酯)添加到含于磷酸钠缓冲液(NaPi,pH 7.4)中的蛋白质的搅拌溶液中。将反应混合物在室温搅拌1小时,然后使用膜过滤浓缩,并在NaPi(pH 8.0)中重新缓冲。将化合物152的NaPi溶液添加到活化蛋白的溶液中,并于rt搅拌16h。然后用无菌水洗涤糖共轭物,并在无菌水中用I-半胱氨酸处理。糖共轭物的纯化通过膜过滤实现。
156的合成
将H-膦酸酯58和接头与吡啶共蒸发,并在真空下干燥30min。之后,将其溶解在吡啶中,并向其中添加PivCl。将反应混合物于rt保持搅拌2h。2h后,将反应冷却至-40℃,添加新鲜制备的I
157的合成
根据化合物33的合成和TBS脱保护步骤进行反应。
157与CRM
根据化合物33的共轭进行反应。
161的合成
针对化合物32的合成描述的过程用于化合物161的合成,此处唯一的变化是在第二步骤中化合物89代替接头用作亲核试剂。
162的合成
其中Z表示
如针对化合物89(除去TBDPS保护基)描述的,然后如针对化合物90描述的那样由化合物161合成化合物162。
163的合成
其中Z表示
如针对化合物89(除去TBDPS保护基)描述的,然后如针对化合物91和92描述的那样由化合物161合成化合物163。
164的合成
其中Z表示
如针对化合物162描述的那样合成膦酸酯化合物164。
165的合成
其中Z表示
如针对化合物163描述的那样合成膦酸酯化合物165。
166的合成
针对化合物86的合成描述的过程用于化合物166的合成。
167的合成
针对化合物96的合成描述的过程用于化合物167的合成。
162的合成
如针对化合物33描述的那样从化合物167合成化合物162。
168的合成
针对化合物60的合成描述的过程用于化合物168的合成。
169的合成
针对化合物86的合成描述的过程用于化合物169的合成。
170的合成
针对化合物87的合成描述的过程用于化合物170的合成。
163的合成
如针对化合物54描述的那样从化合物170合成化合物163。
172的合成
其中Z表示
从连接至十二糖171的十二糖89合成化合物172。
根据针对化合物88描述的过程,如针对化合物89描述的那样遵循TBDPS基团的脱保护,然后如针对化合物90描述的那样进行完全脱保护。
173的合成
其中Z表示
根据针对化合物88描述的过程,如针对化合物89描述的那样遵循TBDPS基团的脱保护,然后如针对化合物92描述的那样进行完全脱保护,从连接到十二糖171的十二糖89合成化合物173。
174的合成
针对化合物96的合成描述的过程用于化合物174的合成。
172的合成
如针对化合物33描述的那样从化合物174合成化合物172。
175的合成
针对化合物60的合成描述的过程用于化合物175的合成。
176的合成
针对化合物86的合成描述的过程用于化合物176的合成。
177的合成
针对化合物87的合成描述的过程用于化合物177的合成。
173的合成
如针对化合物54描述的那样从化合物177合成化合物173。
178的合成
针对化合物96的合成描述的过程用于化合物178的合成。
179的合成
如针对化合物33描述的那样从化合物178合成化合物179。
180的合成
针对化合物60的合成描述的过程用于化合物180的合成。
181的合成
针对化合物86的合成描述的过程用于化合物181的合成
182的合成
针对化合物87的合成描述的过程用于化合物182的合成。
183的合成
如针对化合物54描述的那样从化合物182合成化合物183。
185的合成
其中Z表示
由十八糖161合成化合物185,根据针对化合物89描述的过程从该十八糖161选择性地除去了TBDPS保护基。此后,将TBDPS脱保护的三糖与化合物184反应以便得到糖185。
186的合成
其中Z表示
由糖185合成化合物186,其根据针对化合物89(除去TBDPS保护基)并随后针对化合物90(除去TBDPS保护基)描述的过程转化得到。
187的合成
其中Z表示
由糖185合成化合物187,其根据针对化合物89描述的过程(除去TBDPS保护基)、针对化合物91描述的磷酸化以及随后针对化合物90描述的完全脱保护转化得到。
188的合成
针对化合物96的合成描述的过程用于化合物188的合成。
186的合成
如针对化合物33描述的那样从化合物188合成化合物186。
189的合成
针对化合物60的合成描述的过程用于化合物189的合成。
190的合成
针对化合物86的合成描述的过程用于化合物190的合成。
191的合成
针对化合物87的合成描述的过程用于化合物191的合成。
187的合成
如针对化合物54描述的那样从化合物191合成化合物187。
192的合成
针对化合物96的合成描述的过程用于化合物192的合成。
193的合成
如针对化合物33描述的那样从化合物192合成化合物193。
194的合成
针对化合物60的合成描述的过程用于化合物194的合成。
195的合成
针对化合物86的合成描述的过程用于化合物195的合成。
196的合成
针对化合物87的合成描述的过程用于化合物196的合成。
197的合成
如针对化合物54描述的那样从化合物196合成化合物197。
198的合成
针对化合物96的合成描述的过程用于化合物198的合成。
199的合成
如针对化合物33描述的那样从化合物198合成化合物199。
200的合成
针对化合物60的合成描述的过程用于化合物200的合成。
201的合成
针对化合物86的合成描述的过程用于化合物201的合成。
202的合成
针对化合物87的合成描述的过程用于化合物202的合成。
203的合成
如针对化合物54描述的那样从化合物202合成化合物203。
A.26寡糖205和209的合成
204的合成
针对化合物96的合成描述的过程用于化合物204的合成。
205的合成
如针对化合物33描述的那样从化合物204合成化合物205。
206的合成
针对化合物60的合成描述的过程用于化合物206的合成。
207的合成
针对化合物86的合成描述的过程用于化合物207的合成。
208的合成
针对化合物87的合成描述的过程用于化合物208的合成。
209的合成
如针对化合物54描述的那样从化合物208合成化合物209。
210的合成
针对化合物96的合成描述的过程用于化合物210的合成。
211的合成
如针对化合物33描述的那样从化合物210合成化合物211。
212的合成
针对化合物60的合成描述的过程用于化合物212的合成。
213的合成
针对化合物86的合成描述的过程用于化合物213的合成。
214的合成
针对化合物87的合成描述的过程用于化合物214的合成。
215的合成
如针对化合物54描述的那样从化合物214合成化合物215。
用NaOH裂解化合物33中的磷酸键
接下来,测试和评估了本发明化合物的稳定性。任务是找出化合物33、54、90、92、112、117、162、163、164、165、172和173在配制条件下的稳定程度。在氢氧化铝胶、PBS缓冲液和水中稳定性之前,在室温下将化合物33用0.1M氢氧化钠处理。在此发现,化合物33仅在高度碱性的条件下才非常缓慢地裂解。然而,即使处于如此剧烈的条件下4天(200μL中的10μg33)后,也仅裂解了50%的化合物33,而在HPLC色谱图中仍观察到50%的化合物33是完好无损的(图6和7)。
化合物33在含于PBS中的氢氧化铝胶、PBS和水中的上的稳定性:
接下来,仔细研究化合物33在配制条件下的稳定性。每个配制小瓶中均包含含于i)含于PBS中的氢氧化铝胶或ii)单独的PBS或iii)水(溶液的总体积为500μL)中的30μg33。NaPi在本文中用作PBS的同义词。每个实验使用60μL含0.6mg铝的氢氧化铝胶。将这三种配制的溶液分别置于37℃、25℃和2-8℃达14天。每隔24h时间段,将来自处于37℃、25℃和2-8℃的每个小瓶的i)含于PBS中的氢氧化铝胶,ii)单独的PBS和iii)水的50μL溶液等分,并通过HPLC分析(图8、9、10)。从这些研究中明显可见,化合物33在2℃至37℃的整个温度范围内是稳定的。图8显示了4天后在2-8℃下的稳定性,图9显示了14天后在2-8℃下的稳定性,图10在4天后在25℃下的稳定性,图11显示了14天后在25℃下的稳定性,图12显示了4天后在37℃下的稳定性,以及图13显示了14天后在37℃下的稳定性。
与艰难梭状芽胞杆菌的天然多糖PSII相比,发现化合物33、54、90、92、112、117、162、163、164、165、172和173在上述配制条件下是足够稳定的。
还发现由如下所示的六糖基磷酸酯重复单元组成的艰难梭状芽胞杆菌的天然多糖PSII在NaOH处理中不稳定,在诸如乙酸之类的酸性条件下不稳定,在处于2-8℃、25℃和37℃的溶液中也不稳定。发现在这些条件下,天然PSII迅速降解为不再诱导免疫学作用的降解产物。
因此,以上稳定性实验明确证实,在天然PSII分解成不再可用作疫苗的片段的条件下,本发明的化合物是稳定的,而本文公开的化合物在溶液中是稳定的,不需要冻干和重新溶解,无需冷藏,也不需要使用昂贵的工作冷链系统进行生产和运输。
SDS-PAGE分析.
将样品在微量离心管中混合,并在热循环仪上于95℃加热5min。冷却至室温5min后,将约2.5μg的样品连同10μL的标记物装载到10%聚丙烯酰胺凝胶的相应孔中。样品在120V的恒定电压下运行1h。根据制造说明使用GelCodeTM蓝色安全蛋白染色剂进行染色。将凝胶用去离子水洗涤过夜,并使用凝胶记录系统进行扫描。
糖共轭物的尺寸排阻色谱法(SEC).
通过SEC分析用于免疫研究的糖共轭物,以观察共轭和未共轭的CRM蛋白之间的质量差异。将样品在50mM Tris,20mM NaCl,pH 7,2中稀释,并在装配有Tosoh TSK G2000柱(SWxl,7.8mm x 30cm,5μm)和a Tosoh
糖共轭物的生产
将C.difficile PS-II合成抗原与载体蛋白CRM
糖共轭物36(33-CRM
对用于免疫研究的C.difficile抗原糖共轭物36、56和94进行共轭效率和抗原含量的分析。糖共轭物的MALDI-TOF MS分析显示了良好的共轭效率。共轭和未共轭的CRM
还通过10%SDS-PAGE和SEC分析了糖共轭物,其与未共轭的CRM
研究I–在兔中免疫的C.diff抗原PS-II的半合成糖共轭物的免疫学评估。
1.研究目的:
评估用C.diff抗原PS-II半合成CRM
2.材料:
·ELISA板(高结合力,EIA/RIA板,96孔,带低蒸发盖的平底,公司:
·检测抗体:山羊抗兔IgG过氧化物酶共轭物(Sigma,#A4914)
·封闭溶液:1%FCS于PBS中(v/v)
·抗体稀释剂:PBS+1%BSA(w/v).
·洗涤缓冲液:PBS+0.1%吐温20(PBS-T)
·显影液:1step
·终止溶液:2M硫酸(H
·酶标仪:Anthos ht 2.
·软件:用于吸光度测量的WinRead 2.36以及用于数据绘图和分析的GraphPadPrism.
·不完全弗氏佐剂(IFA).InvivoGen;Cat:vac-ifa-10,批次号:IFA-39-03;到期日:2019年9月
·QuantiPro
3.方法:
用于免疫的疫苗的配制
将C.difficile PS-II糖共轭物36配制在不完全弗氏佐剂(IFA)中以在兔中免疫。使用来自Invivogen的不完全弗氏佐剂(IFA)来配制用于兔免疫研究的疫苗。按照制造规程。抗原:将IFA浓度保持在1:1。每只动物的抗原剂量保持在2.5μg/200μL/只动物(100μL抗原+100μL IFA)。将所需计算体积(最终免疫体积的50%)的IFA放入15mL无菌falcon中。将计算量的稀释抗原溶液(用PBS将体积调节至最终免疫体积的50%)放入装配有20G针头的3mL无菌注射器中。将DS溶液添加到含有IFA的falcon中,并立即涡旋15秒钟(5X)。制剂的颜色在涡旋时从浅黄色变为乳白色,这表明形成了稳定的乳液。将得到的疫苗制剂短暂涡旋并等分到具有所需剂量体积的2mL无菌管中。免疫前,将含有疫苗制剂的管涡旋,然后注射入动物体内。
免疫时间表
在无特定病原体的条件下进行兔免疫,并随意提供食物和水。用疫苗制剂以200μL/兔的注射量对兔(n=4)进行皮下免疫。用于兔的抗原剂量保持为每只动物2.5μg的PS-II抗原或相应体积的PBS用于阴性对照。在第0、14和35天对兔进行免疫。在第0、7和42天抽血以确定抗体滴度。
4.使用内部抗原涂覆板进行血清的酶联免疫吸附测定(ELISA)
用抗原涂覆板
抗原-BSA共轭物用作涂覆抗原。将抗原-BSA共轭物以5μg/mL的浓度溶解在pH 7.4的磷酸盐缓冲盐水(PBS)中。每孔涂覆100μL,并于4℃温育过夜,以获得0.5μg/孔的抗原浓度。
洗涤
抗原吸附过夜后,将板用PBS-T(200μL/孔)洗涤1X,并通过倒转板并用干净的干纸巾(tissue towel)轻拍除去每孔多余的液体。
封闭
用200μL的商业封闭溶液将板封闭,并于rt温育2h。
洗涤
封闭后,将板用PBS-T(200μL/孔)洗涤3X,并通过倒转板并用干净的干纸巾轻拍除去每孔多余的液体。
血清的稀释和温育
将来自不同实验组的不同时间点的合并血清(n=4只兔子)在抗体稀释剂(PBS+1%BSA)中稀释至它们各自的稀释度。将不同实验组的100μL的稀释血清样品一式两份添加到相应的孔中,并在设定为250rpm的振荡器上于rt温育2h。100μL/孔的抗体稀释液(PBS+1%BSA)形成实验空白。与血清一起温育后,将板用PBS-T(200μL/孔)洗涤4X,并通过倒转板并用干净的干纸巾轻拍除去每孔多余的液体。
温育(检测抗体)
将相应的检测抗体抗兔IgG HRP共轭物在抗体稀释液(PBS+1%BSA)中以1:10,000的比例稀释,并以100μL/孔添加,并在振荡器上以250rpm于rt温育1h。与检测抗体一起温育后,将板用PBS-T(200μL/孔)洗涤5X,并通过倒转板并用干净的干纸巾轻拍除去每孔多余的液体。
底物添加
向每个孔中添加100μL的随时可用的TMB底物(从4℃归一化为RT),并在黑暗中温育15min。通过添加50μL/孔的2M H
结果
通过使用Graphpad Prism软件绘制曲线图来分析吸光度。
ELISA数据清楚地表明,来自经C.difficile PS-II共轭物36免疫的兔的血清可以识别相应的抗原(见图15)。
研究II–在兔和小鼠中免疫的C.diff抗原PS-II的半合成糖共轭物的免疫学评估.
1.研究目的:
评估用C.diff PS-II半合成CRM
2.材料
·ELISA板(高结合力,EIA/RIA板,96孔,带低蒸发盖的平底,公司:
·检测抗体:山羊抗兔IgG过氧化物酶共轭物(Sigma,#A4914)和抗人IgG(H+L)-HRP,Nordic Immunology,批号:6276
·封闭溶液:Roche,Ref:11112589001;批号:21495200,到期日:2019年7月.
·抗体稀释剂:PBS+1%BSA(w/v)
·洗涤缓冲液:PBS+0.1%吐温20(PBS-T)
·显影液:1step
·终止溶液-2M硫酸(H
·酶标仪:Anthos ht 2.
·软件:用于吸光度测量的WinRead 2.36以及用于数据绘图和分析的GraphPadPrism
·明矾:氢氧化铝凝胶佐剂(氢氧化铝胶2%),Brenntag,批次号:5447到期日:2020年2月
·QuantiPro
·
·Precision Plus Dual Color,Cat:1610374;对照号:641798899
·Gel Code
·用于菌株630的C.diff涂覆的ELISA板(tgc BIOMICS批号:630-43411)和R20291(tgc BIOMICS批号:R20291-43559)到期日:05/2020.
·C.diff阳性患者血浆。
3.方法
在氢氧化铝(明矾)佐剂中配制用于免疫的疫苗
所有制剂均在无菌条件下制备。将糖共轭物56和94(药物物质;DS)和PBS以适当的预先计算的比例混合在50mL Falcon
免疫时间表
在无特定病原体的条件下进行小鼠和兔免疫,并随意为动物提供食物和水。用疫苗制剂,用不同的抗原剂量以100μL/只小鼠和500μL/只兔的注射体积对小鼠(每个研究组7或8只兔)和兔(每个研究组n=4)进行皮下免疫。在第0、14和28天对小鼠进行免疫,并在第21和35天采血。在0、14、28和77天对兔进行免疫,并在第0、7、21、35、77和84天采血。从血液样品中制备血清用于血清抗体分析。
4.使用内部抗原涂覆的板进行血清的酶联免疫吸附测定(ELISA).
用抗原涂覆板:
将共轭物54-BSA和92-BSA用作涂覆抗原。在pH 7.4的磷酸盐缓冲盐水(PBS)中将相应共轭物稀释至5μg/mL的浓度。每孔涂覆100μL,并于4℃温育过夜,以获得0.5μg/孔的抗原浓度。为了涂覆分离的PS-II多糖,将多糖在具有10mM咪唑的PBS中稀释至50μg/mL,并且于50℃以100μL/孔涂覆5h。
洗涤:
吸附抗原后,将板用PBS-T(200μL/孔)洗涤1X,并通过倒转板并用干净的干纸巾轻拍除去每孔多余的液体。
封闭:
用200μL商业封闭溶液封闭板,并于rt温育2h。
洗涤:
封闭后,将板用PBS-T(200μL/孔)洗涤3X,并通过倒转板并用干净的干纸巾轻拍除去每孔多余的液体。
将来自不同实验组的不同时间点的合并血清(n=4只兔或n=7-8只小鼠/组)在抗体稀释剂(PBS+1%BSA)中稀释至它们各自的稀释度。将不同实验组的100μL的稀释血清样品一式两份添加到相应的孔中,并在设定为250rpm的振荡器上于rt温育2h。为了进行竞争ELISA实验,将稀释的血清与10或50μg的分离的PS-II多糖或与PBS一起在冰上温育30min,然后添加到ELISA板中。100μL/孔的抗体稀释液(PBS+1%BSA)形成实验空白。与血清一起温育后,将板用PBS-T(200μL/孔)洗涤4X,并通过倒转板并用干净的干纸巾轻拍除去每孔多余的液体。
将相应的检测抗体,抗兔或抗小鼠IgG HRP共轭物在抗体稀释液(PBS+1%BSA)中以1:10,000的比例稀释,并以100μL/孔添加,并在振荡器上以250rpm于RT温育30min。与检测抗体一起温育后,将板用PBS-T(200μL/孔)洗涤5X,并通过倒转板并用干净的干纸巾轻拍除去每孔多余的液体。
底材添加:
向每个孔中添加100μL的随时可用的TMB(3,3’,5,5’-四甲基联苯胺)底物(从4℃归一化为RT),并在黑暗中温育15min。通过添加50μL/孔的2M H
结果:
通过使用GraphPad Prism软件绘制图表来分析吸光度。
5.使用商业预涂板进行血清的酶联免疫吸附测定(ELISA)
除了省去了涂覆步骤之外,该过程与上述ELISA方案相同。
结果:
来自免疫兔的血清IgG识别免疫原(图20),分离的PS-II多糖(图19B和19C)和C.difficile菌株630(图16和17),R20291(图18)和VPI10463(图19A)。来自免疫小鼠的血清IgG识别相应的免疫原(图23)和C.difficile菌株630(图21)和R20291(图22)。
本文提供的数据证实,在用本发明的共轭物,特别是共轭物56和94免疫后,在兔和小鼠体内引起针对本发明的寡糖以及针对天然C.difficile PS-II多糖(分离并在该细菌的表面上)的功能性抗体。这些发现表明这些抗体具有赋予免受C.difficile感染的保护性的潜力。
ELISA数据进一步证明本发明的共轭物是免疫原性的并且诱导高抗体滴度。因此,ELISA分析表明,本发明的糖在兔和小鼠体内是免疫原性的,并产生交叉反应的抗体。
- 抗艰难梭状芽胞杆菌的稳定疫苗
- 用于艰难梭状芽胞杆菌感染的4,6-二(邻硫代磷酸盐)-肌醇-1,2,3,5-四-邻硫酸盐