一种孟鲁司特钠中间体的合成方法
文献发布时间:2023-06-19 18:37:28
技术领域
本发明涉及一种孟鲁司特钠中间体的合成方法,属于药物中间体制备领域。
背景技术
孟鲁司特钠(Monte lukast Sodium),化学名称为1-[[[(1R)-1-[3-[(1E)-2-(7-氯-2-喹啉)乙烯基]苯基]-3-[2-(1-羟基-1-甲基乙基)苯基]丙基]硫代]甲基]环丙烷乙酸钠,是一种白三烯受体拮抗剂,可抑制白三烯的生物合成,在药学上可用作止喘剂、抗过敏剂等。
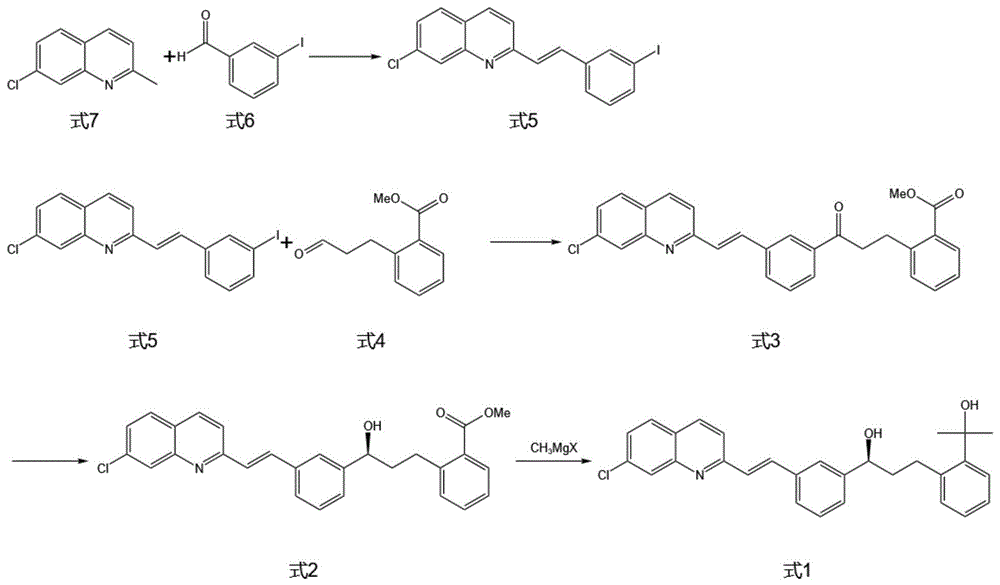
式1化合物2-[3-(S)-[3-(2-(7-氯-2-喹啉基)乙烯基)苯基]-3-羟丙基]苯基-2-丙醇是制备孟鲁司特钠的重要中间体,其化学结构式如下所示:
在专利CN101638381A中,公开了孟鲁司特钠中间体的合成方法,使用 3-氰基苯甲醛和7-氯喹哪啶进行缩合得到中间化合物3-(2-(7-氯-2-喹啉基)乙烯基)苯甲氰,再与化合物2-(2-邻(2-卤乙基)苯基丙基)四氢吡喃醚反应生成最终产物2-(2-(3-(2-(7-氯-2-喹啉基)乙烯基)苯基)-3-氧代丙基)苯基)丙醇,该反应路线步骤较多,生产流程长,其合成路线如下所示:
在专利WO2008035086中,公开了以(S)-1-(3-溴苯基)-3-[2-(1-羟基-1-甲基乙基)-苯基]甲酸甲酯为原料制备孟鲁司特钠中间体的合成方法,该法在将酯基转变成叔醇基团时,转化率较低,且整体收率与纯度都不高,导致生产成本较高,不利于工业化生产,其制备路线如下:
从以上文献中可以看出其共同存在的缺点是反应步骤多,生产流程长,产率及产物纯度较低。生产成本高。
因此,目前亟需寻找一种反应路线短、产物率高、产品纯度高的合成方法。
发明内容
本发明针对以上现有技术中存在的缺陷,提供一种孟鲁司特钠中间体的合成方法,解决的问题是如何实现提高产品纯度的制备方法。
本发明的目的是通过以下技术方案得以实现的,一种孟鲁司特钠中间体的合成方法,该方法包括以下步骤:
S1:在有机溶剂中,使式7化合物与式6化合物发生缩合反应,生成式5 的化合物;
S2:在有机溶剂中式5化合物与式4化合物在催化剂与相转移催化剂的作用下反应生成式3化合物;
S3:式3化合物在还原剂的催化下发生选择性还原反应生成式2化合物;
S4:式2化合物与催化剂反应生成最终产物。
式5化合物合成路线为:
式1化合物合成路线为:
其中,卤素X为氯。
本发明通过7-氯喹哪啶与3-碘苯甲醛在高温下缩合得到中间产物式5化合物,所有反应原料都能够直接从外界获得,无需单独制备,节省了合成原料的时间与成本,再将式5化合物与式4化合物2-(3-氧代丙基)苯甲酸甲酯反应生成式3化合物,使用还原剂选择性还原式3化合物,再将得到的式2化合物与甲基卤化镁反应生成最终产物,整体反应路线较为简短,无需消耗大量的人工制备中间产物及各种原料,缩短了生产流程,产率高与产物纯度大,同时反应后还可对部分副产物进行回收制备反应原料再次利用,减少了生产成本。
在上述孟鲁司特钠中间体的合成方法中,作为优选,所述S1步骤中有机溶剂选用乙酸酐、三氟乙酸酐或醋酐其中一种。作为最优选,乙酸酐作为反应溶剂时溶解度最大。
在上述孟鲁司特钠中间体的合成方法中,作为优选,所述S2步骤中有机溶剂先用四氢呋喃、甲苯或二氧六环其中一种。作为最优选,加入四氢呋喃反应速度最快。
在上述孟鲁司特钠中间体的合成方法中,作为优选,所述S2步骤中催化剂可选用醋酸钯、三氟乙酸钯或钯碳其中一种。作为最优选,使用醋酸钯作为催化剂时能够减少副产物的生成,降低生产成本。
在上述孟鲁司特钠中间体的合成方法中,作为优选,所述S2步骤中相转移催化剂可选用苄基三乙基溴化铵、四丁基溴化铵或四丙基溴化铵其中一种。作为最优选,选用苄基三乙基溴化铵时产物纯度较高。
在上述孟鲁司特钠中间体的合成方法中,作为优选,所述S2步骤中反应在碱性环境下发生,可选用三乙胺、六氢吡啶或吡咯烷。作为最优选,选用三乙胺时副产物生成少,产物纯度较高。
在上述孟鲁司特钠中间体的合成方法中,作为优选,所述S3步骤中还原剂选用(-)-二异松蒎基氯硼烷。
在上述孟鲁司特钠中间体的合成方法中,作为优选,所述S4步骤中催化剂为有机钛试剂。作为最优选,使用无水四氯化钛配置有机钛试剂。
在上述孟鲁司特钠中间体的合成方法中,作为优选,所述S4步骤中反应溶剂为甲苯或二甲苯。作为最优选,选用甲苯时产物收率较大。
在上述孟鲁司特钠中间体的合成方法中,作为优选,所述S3步骤结束后可将溶液中的碘进行回收制备式6化合物,能够减少生产成本,降低后处理难度。
综上所述,本发明与现有技术相比,具有以下优点:
1.本发明中生产原料皆可从外界购买获得,无需消耗人力物力单独制备,同时部分原料在反应结束后能够回收再利用,能够减少生产成本,降低后处理难度;
2.本发明反应路线的转化率较高,因此产物生成率较高,反应中多数为催化剂作用下的定向反应,因此副产物较少,产物纯度较高;
3.本发明中未使用大量剧毒化合物如乙腈等,同时严格控制反应条件,减少反应过程中产生对工作人员有害的物质,安全性较高。
附图说明
图1为本发明总合成路线;
图2为本发明式1化合物结构式。
具体实施方式
下面通过具体实施例,对本发明的技术方案作进一步具体的说明,但是本发明并不限于这些实施例。
实施例1
式5化合物的制备:
将44.4g式7化合物(0.25mol)和37.53g式6的化合物(0.25mol)溶于300mL 乙酸酐中,加热至70℃,搅拌反应24h,减压浓缩,500mL乙酸乙酯萃取3 次,过滤,依次使用200mL饱和食盐水溶液及200mL乙酸乙酯洗涤滤饼,加入20g无水硫酸钠干燥,得到67.29g式5的化合物,收率86.9%,纯度为98.1%。
实施例2
式3化合物的制备:
将37.27g式5化合物(0.1mol)、29.9g式4化合物(0.18mol)和38.11g苄基三乙基溴化铵(0.14mol)、12g无水硫酸镁(0.1mol)溶于四氢呋喃(300mL)中,在氮气保护下向所得的溶液中加入4.5g醋酸钯(0.02mol)和13.15g三乙胺 (0.13mol),将溶液加热到50℃,搅拌反应4小时,加入50mL水,200mL乙酸乙酯萃取3次,200mL饱和氯化钠洗涤,浓缩,柱层析纯化(石油醚:乙酸乙酯=10:1),浓缩得到40.8g式3化合物收率89.5%,纯度为95.3%。
实施例3
式2化合物的制备:
在反应瓶中,将45.59g式3化合物(0.1mol)溶于500mL二氯甲烷中,加入32mLN,N-二异丙基乙胺,降温至-15℃搅拌反应30min,缓慢滴加148mL(-)- 二异松蒎基氯硼烷正庚烷溶液,滴毕,保温反应8h,反应结束后升温至25℃,加入25mL三乙胺搅拌2h,加入饱和氯化钠溶液500mL,静置分层,300mL 二氯甲烷萃取3次,合并有机相,200mL饱和盐水洗涤,蒸出溶剂,加入250mL 甲苯,55℃溶清后冷却至室温搅拌12h,过滤,滤饼用50mL正庚烷洗涤2次,50℃减压干燥,得40.85g式2化合物,收率为89.2%,纯度为98.8%。
实施例4
式1化合物的制备:
氮气保护下,在反应瓶中加入18.96g无水四氯化钛(0.1mol)和50mL四氢呋喃,降温至0℃,滴加30mL3M甲基氯化镁四氢呋喃溶液,控温15℃反应 20min,得有机钛试剂。
在另一反应瓶中加入45.8g式2化合物(0.1mol)和200mL甲苯,搅拌至溶清。将有机钛试剂滴加至溶液中,滴加完毕后,0-10℃反应5h。反应液中加入200mL醋酸氨水溶液静置分层,有机相依次使用200mL10%碳酸钠溶液,200mL5%亚硫酸钠水溶液和200mL饱和食盐水冲洗。加入90mL乙醇,搅拌析晶2h,抽滤,干燥,生成43.65式1化合物,收率为95.1%,纯度为99.3%。
实施例5
碘的回收与3-碘苯甲醛的制备:
往反应瓶中加入20.1g三乙胺氢碘酸盐,加100mL水溶解,过滤,滤液中加入氢氧化钾溶液调节pH至10,二氯甲烷萃取三乙胺及其他有机杂质,分出水相,浓缩,加入100mL乙醇降温析晶,生成9g碘化钾。滴定分析,总回收率89%。
三口瓶中加入7.66g间氨基苯甲醛和25mL浓盐酸,100mL水,冰浴冷却搅拌,滴加3.9g亚硝酸钠溶液,温度不高于5℃,滴毕,继续搅拌反应2小时后得重氮盐溶液。将9.28g碘化钾缓缓加入到以上重氮盐溶液中,继续反应 30min,抽滤,水洗,干燥,生成10.0g3-碘苯甲醛,总收率为76%。
实施例6
将44.4g式8化合物7-氯喹哪啶和37.53g式7化合物3-羧基苯甲醛溶于 500mL三氟乙酸酐中,加热至50℃~140℃,搅拌反应24h,减压蒸出乙酸酐溶剂,500mL乙酸乙酯萃取3次,过滤,使用200mL饱和食盐水溶液和mL 乙酸乙酯洗涤滤饼,加入20g无水硫酸钠干燥,真空浓缩,得到64.03g式6 的化合物,收率82.7%,纯度为96.6%。
实施例7
将44.4g式8化合物7-氯喹哪啶和37.53g式7化合物3-羧基苯甲醛溶于 500mL醋酐中,加热至50℃~140℃,搅拌反应24h,减压蒸出乙酸酐溶剂, 500mL乙酸乙酯萃取3次,过滤,使用200mL饱和食盐水溶液和mL乙酸乙酯洗涤滤饼,加入20g无水硫酸钠干燥,真空浓缩,得到65.5g式6的化合物,收率84.6%,纯度为97.2%。
实施例8
将37.27g式5化合物、29.9g式4化合物和38.11g四丁基溴化铵、12g无水硫酸镁溶于300mL甲苯中,在氮气保护下向所得的溶液中加入6.45g三氟乙酸钯和11.07g六氢吡啶,将溶液加热到50℃,搅拌反应4小时,加入50mL 水,200mL乙酸乙酯萃取3次,200mL饱和氯化钠洗涤,浓缩,柱层析纯化(石油醚:乙酸乙酯=10:1),浓缩得到39.61g式3化合物收率86.9%,纯度为94.1%
实施例9
将37.27g式5化合物、29.9g式4化合物和37.28g四丙基溴化铵、12g无水硫酸镁溶于300mL二氧六环中,在氮气保护下向所得的溶液中加入2.13g 钯碳和9.25g吡咯烷,将溶液加热到50℃,搅拌反应4小时,加入50mL水, 200mL乙酸乙酯萃取3次,200mL饱和氯化钠洗涤,浓缩,柱层析纯化(石油醚:乙酸乙酯=10:1),浓缩得到38.84g式3化合物收率85.2%,纯度为91.7%
实施例10
氮气保护下,在反应瓶中加入18.96g无水四氯化钛和50mL四氢呋喃,降温至0℃,滴加30mL3M甲基氯化镁四氢呋喃溶液,控温15℃反应20min,得有机钛试剂。
在另一反应瓶中加入45.8g式2化合物和200mL二甲苯,搅拌至溶清。将有机钛试剂滴加至溶液中,滴加完毕后,0-10℃反应5h。反应液中加入 200mL醋酸氨水溶液静置分层,有机相依次使用200mL10%碳酸钠溶液, 200mL5%亚硫酸钠水溶液和200mL饱和食盐水冲洗。加入90mL乙醇,搅拌析晶2h,抽滤,干燥,生成42.36式1化合物,收率为92.3%,纯度为98.1%。
对比例
本实施例为已公开专利CN102442948B的实施例
式III化合物2-(2-(3(S)-氯-3-(3-(2-(7-氯-2-喹啉基)-(E)乙烯基)苯基)丙基)苯基)-2-丙醇的制备方法:
在室温(25-30℃)以及氩气保护下,在反应瓶中依次加入2-(2-(3(R)-羟基-3-(3-(2-(7-氯-2-喹啉基)-(E)乙烯基)苯基)丙基)苯基)-2-丙醇(25.1g,55mmol)、 DMF(400mL)和二异丙基乙基胺(17.74g,137.5mmol),搅拌至完全溶解。冰盐浴将体系降温至-10℃,于30分钟内滴加甲磺酰氯(15.6g,137.5mmol),然后于该温度下反应10小时。(TLC或者HPLC监测原料反应完毕);一次性加入氯化锂(11.7g,275mmol),撤去冰浴,自然回至室温,继续反应14小时。TLC 跟踪反应完毕。加入冰水300mL乙酸乙酯(60mL×3),合并有机相。依次用饱和食盐水,水洗涤有机相。无水硫酸钠干燥。浓缩有机相到原体积的1/5,加入石油醚500mL搅拌半小时,然后静置过夜,过滤,干燥得16.1g式III化合物白色固体,收率61.5%
对比本发明中的制备方法,此实施例中收率较低,导致生产成本偏高,不利于大规模生产。
本发明的实施方式并不限于上述实施例所述,在不偏离本发明的精神和范围的情况下,本领域普通技术人员可以在形式和细节上对本发明做出各种改变和改进,而这些均被认为落入了本发明的保护范围。