一种接枝型聚醚大单体及其在聚羧酸减水剂中的应用
文献发布时间:2024-04-18 19:58:26
技术领域
本发明涉及混凝土外加剂技术领域,具体涉及一种接枝型聚醚大单体及其在聚羧酸减水剂中的应用。
背景技术
基作为第三代高性能减水剂,聚羧酸减水剂由直线型的聚乙二醇侧链及含羧基等吸附基团的主链组成,其梳形拓扑结构使聚羧酸具有结构易设计、高减水率、高保坍性等优点,因而被广泛应用于混凝土生产。
粘土矿物主要由高岭石、伊利石和蒙脱石中一种或多种组成,其中蒙脱石具有易吸水膨胀的特性,且容易与聚羧酸中聚乙二醇侧链形成插层结构。蒙脱土与水泥颗粒对聚羧酸的竞争吸附作用正是聚羧酸工作性能显著降低的主要因素。为抑制粘土矿物对聚羧酸工作性能的不利影响,目前采取的措施主要包括水洗骨料、掺加过量减水剂、采用粘土改性剂与聚羧酸减水剂复配以及功能单体改性聚羧酸分子结构。
专利CN107043227B公开了一种由羧基或磷酸基团取代芳环化合物、端芳环聚乙二醇以及醛类化合物合成的抗粘土型聚合物分散剂,可以与聚羧酸减水剂复配使用,提高减水剂的抗粘土性能。然而,该聚合物分散剂的合成体系以硫酸为反应介质,且反应温度较高,生产危险系数较高,阻碍了其大规模工业生产。
专利CN109957076B公开了一种抗泥保坍型聚羧酸减水剂及其制备方法,由聚醚单体依次与异氰酸糠酯、N-氨基甲酰马来酰亚胺进行封端反应,合成功能性封端聚醚单体,随后该单体与丙烯酸聚合制备聚羧酸减水剂。由于所制备聚醚单体末端环形结构阻止聚羧酸侧链与粘土晶层间插层结构的形成,而提高了聚羧酸减水剂的分散能力和保坍性能。然而,功能性聚醚单体反应条件较为苛刻,合成步骤繁琐,生产成本较高,并不利于其工业生产应用。
专利CN112876619A公开了一种聚合物侧链含有胍环大环官能团的聚羧酸减水剂,对比常规梳形聚羧酸减水剂,胍环官能团的存在可增大聚羧酸的空间位阻效应,减少粘土矿物导致的聚羧酸的无效吸附。接枝胍环大单体的合成浓度较低且要求较长反应时间,生产成本较高,限制了该功能单体的工业生产。
专利CN113072667A公开了一种由不饱和酸单体A、直链聚醚单体B与支链聚醚单体C进行自由基共聚反应得到所述新型聚羧酸减水剂。该减水剂具有减水率大,优良的抗泥功能。该聚羧酸减水剂中支链聚醚的合成要求多步反应,且其聚合基团活性较低较难聚合。
虽然对聚羧酸侧链引入功能基团或对聚羧酸聚醚侧链末端官能团改性等技术方法,在抑制粘土矿物对聚羧酸减水剂分散性能方面取得一定的成效,但均未能根本上解决聚羧酸侧链在粘土矿物晶层的插层吸附问题。因此,从粘土矿物对聚羧酸减水剂的吸附作用机理出发,设计合成具备高减水率且能够有效避免聚羧酸侧链插层吸附现象的高性能减水剂,提高减水剂的抗粘土性能,对于拓宽聚羧酸减水剂的应用范围,提高其混凝土骨料适应性具有十分重要的意义。
发明内容
为了解决现有的技术中聚羧酸减水剂对混凝土骨料中粘土矿物敏感度高、分散性差的问题,本发明提供接枝型聚醚大单体及其在聚羧酸减水剂中的应用,该接枝型聚醚大单体改变了传统聚醚大单体的直线型结构,并应用于聚羧酸减水剂中,改变了传统聚羧酸减水剂聚醚侧链的直线型拓扑结构,从根本上降低了聚羧酸减水剂对粘土矿物的敏感性,提升其对砂石骨料的适应性。
一种接枝型聚醚大单体,上述接枝型聚醚大单体由端羟基类不饱和化合物和环氧化合物开环聚合反应得到;上述接枝型聚醚大单体结构如下式(1)所示:
其中,100≥k≥0,100≥n≥0,100≥m>0;R
优选的,5≥k≥1,30≥m≥5,25≥n≥15;
优选的,R
优选的,接枝型聚醚大单体包括式(I-1)-(I-6)所示化学结构中的至少一种;
上述接枝型聚醚大单体的制备方法,包括以下步骤:
S1a.将端羟基类不饱和化合物进行干燥处理;
S2a.将端羟基类不饱和化合物置于聚合釜中,加有机溶剂稀释后进行除氧操作,并恒温至聚合温度;向体系中加入聚合催化剂,缓慢加入环氧化合物进行聚合反应;
S3a.聚合反应后调节聚合体系至中性,随后过氧化铝层析柱以除去残余催化剂,所得聚合物在有机溶剂中溶解后在乙醚中沉降并真空干燥,得到接枝型聚醚大单体。
上述步骤S1a的干燥温度为80~150℃;上述步骤S1a的干燥时间为2~10h。
优选的,干燥温度为90~110℃;干燥时间为3~5h。
上述步骤S2a中端羟基类不饱和化合物与催化剂的摩尔比为1:(1-3)。
根据支链聚醚的设计分子量不同,端羟基类不饱和化合物与聚合单体环氧化合物的摩尔比为1:(5-20)。
上述步骤S2a的反应温度为40~70℃;上述步骤S2a的反应时间为10~24h。
优选的,步骤S2a的反应温度为50~60℃,步骤S2a的反应时间为15~20h。
上述步骤S2a的有机溶剂为甲苯、二氯甲烷、四氢呋喃、二甲基亚砜或N,N-二甲基甲酰胺中的至少一种。
上述步骤S2a的聚合催化剂选自阳离子催化剂或阴离子催化剂;上述阳离子催化剂为强质子酸或路易斯酸中的至少一种;上述强质子酸为硫酸、磺酸、高氯酸中的至少一种;上述路易斯酸为三氯化铁、四氯化锡中的至少一种;上述阴离子催化剂为无机碱、有机碱或膦腈碱中的至少一种;上述无机碱为氢化钠、氢化钾、氢氧化钾、氢氧化钠、氢氧化锂中的至少一种;上述有机碱为四甲基氢氧化铵、四丁基氢氧化磷中的至少一种;上述膦腈碱为叔辛基亚氨基-三(二甲氨基)正膦、叔丁基亚氨基-三(二甲氨基)正膦、叔丁基亚氨基-三(吡咯烷)膦、亚氨基-三(二甲氨基)磷酸中的至少一种。
上述步骤S3a的有机溶剂为正戊烷、环戊烷、正己烷、环己烷、正庚烷、石油醚中的至少一种。
接枝型聚醚大单体的应用,上述接枝型聚醚大单体用于制备聚羧酸减水剂。
一种聚羧酸减水剂,该聚羧酸减水剂包括结构单元A、结构单元B、结构单元C,上述结构单元A、结构单元B、结构单元C的摩尔比为(0-30):(0-30):(40-100);上述结构单元A的来源单体为本发明上述的接枝型聚醚大单体,结构如下式(1-1)所示:
其中100≥k≥0,100≥n≥0,100≥m>0;R
优选的,5≥k≥1,30≥m≥5,25≥n≥15;
上述结构单元B的结构如下式(2)所示:
其中,100≥r>0;R
优选的,50≥r≥30;
上述结构单元C的结构如下式(3)所示:
其中,M选自H
优选的,上述结构单元A、结构单元B、结构单元C的摩尔比为(5-30):(0-30):(50-75)。
上述结构单元A的来源单体为权利要求1上述的接枝型聚醚大单体。
上述结构单元B的来源单体为不饱和聚醚单体,上述不饱和聚醚单体为烯丙基聚氧乙烯醚、异戊烯基聚氧乙烯醚、(甲基)烯丙基聚氧乙烯醚、(甲基)丙烯酸聚氧乙烯酯中的至少一种;上述不饱和聚醚单体的数均分子量Mn为400-5000g/mol。
上述结构单元C的来源单体为丙烯酸、甲基丙烯酸、马来酸、衣康酸、丁烯酸或上述酸的盐或酸酐中的至少一种。
一种聚羧酸减水剂的制备方法,包括以下步骤:(1)向反应装置中加入不饱和聚乙二醇单体并加水溶解得均匀体系;(2)聚合体系升温至特定温度,在搅拌过程中滴加不饱和酸、所制备接枝聚醚单体、链转移剂、水和引发剂的混合溶液,滴加时间1~4小时,滴加完毕继续反应2~4小时;(3)加入碱性水溶液,调节聚合体系pH至中性,随后冷却至室温。
上述步骤(2)的聚合温度为20℃~60℃。
上述氧化剂选自过氧化氢、过硫酸钾、过硫酸铵、过氧化氢中的至少一种;还原剂选自硫酸亚铁、亚硫酸钠、抗坏血酸、烷基叔胺、硫醇中的至少一种;上述氧化剂和还原剂的总用量为共聚单体总用量的0.1%~1%,其中氧化剂与还原剂的摩尔比为1:(1-2)。
上述链转移剂为异丙醇、亚硫酸氢钠、甲酸钠、次磷酸钠、巯基乙醇、巯基乙酸、巯基丙醇或巯基丙酸中的至少一种;链转移剂总用量为共聚单体总用量的0.01%~2%。
一种聚羧酸减水剂的应用,上述聚羧酸减水剂应用于水泥基材料中,且其折固掺量为胶凝材料用量的0.1%~0.5%。
本发明相比现有技术具有以下优点:
1、本发明从聚羧酸减水剂分子拓扑结构及其与蒙脱土晶层间作用机理出发,设计合成具备接枝侧链结构的聚醚单体及相应聚羧酸减水剂,改变传统聚羧酸减水剂聚醚侧链的直线型拓扑结构,从根本上降低聚羧酸减水剂对粘土矿物的敏感性,提升聚羧酸减水剂对砂石骨料的适应性,相较于传统聚羧酸减水剂在蒙脱土存在下对水泥具有更好的吸附分散性和减水保坍性;
2、本发明所合成接枝型聚醚单体以及相应的聚羧酸减水剂的结构可设计性高,通过调节接枝型聚醚单体的用量调控接枝结构及接枝密度,可有效改善接枝型聚醚以及相应聚羧酸减水剂的物化性质,为砂石骨料的适应性调控提供更大优化空间;
3、本发明接枝型聚醚单体以及相应的聚羧酸减水剂的生产工艺与传统聚羧酸减水剂生产工艺相类似,生产线升级改造容易,且合成工艺简单成本较低。
附图说明
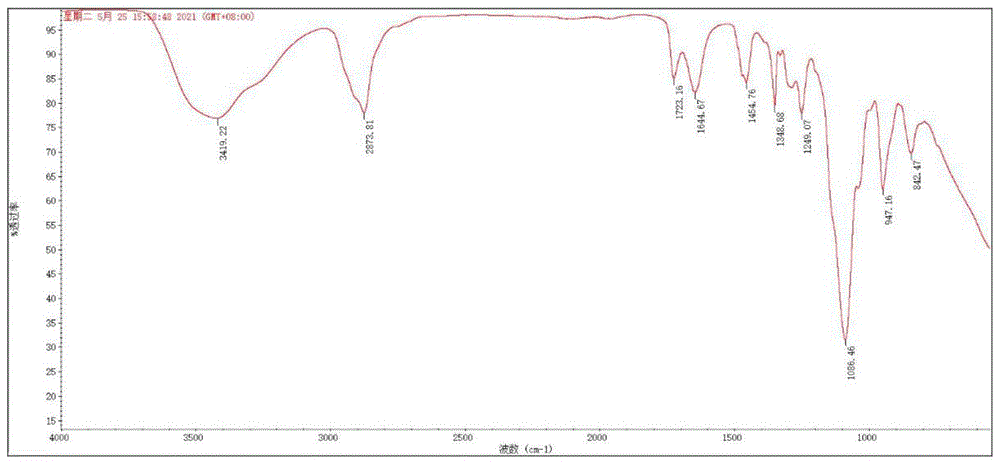
图1为实施例1接枝型聚醚型聚羧酸减水剂PCAP-1的FT-IR谱图;
具体实施方式
下面将结合本发明实施例,对本发明的技术方案进行清楚、完整的描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。实施例中所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。本发明实施例中,接枝聚醚及聚羧酸减水剂的数均分子量Mn、重均分子量Mw和分子量分布系数PDI等参数由Wyatt technology corporation凝胶渗透色谱仪测定。采用Shodex SB806+803凝胶渗透色谱柱串联,检测器为Shodex RI-7型示差折光检测器,以0.1M NaNO3溶液为洗提液,其流动相速度为0.8ml/min,窄分布聚乙二醇为GPC标样,对聚合物流出分子量校准。以DMSO-d
接枝型聚醚大单体I-1的合成
该实施例中以数均分子量Mn=1000kg/mol的聚乙二醇甲基丙烯酸酯(MPEG1000)为环氧化合物单甲基三氧乙烯基缩水甘油醚(TEOG)开环聚合端羟基类不饱和化合物。
端羟基类不饱和化合物MPEG100预处理:将MPEG1000(含600ppm对羟基苯甲醚阻聚剂)置于100℃真空烘箱中干燥4小时后冷却至室温,以除去MPEG1000中残余水分;
环氧单体TEOG聚合:于氮气气氛下,将干燥的开环聚合端羟基类不饱和化合物MPEG1000(1.8g,1.80mmol)以及四正丁基氯化铵(20.6mg,0.072mmol)置于聚合釜中,加入3mL甲苯溶解。将反应体系恒温至10℃,随后向体系中加入聚合催化剂三异丁基铝(0.7mL,0.77mmol),通过蠕动泵缓慢加入环氧单体TEOG(4.21g,18.00mmol),加料6小时随后在该温度下继续反应10小时。单体转化率由
1
接枝型聚醚大单体I-3的合成
该实施例中以甲基丙烯酸羟丙酯(HPMA)为环氧化合物单甲基三氧乙烯基缩水甘油醚(TEOG)开环聚合端羟基类不饱和化合物。
环氧乙烷(EO)及环氧单体TEOG聚合:于氮气气氛下,将干燥的开环聚合端羟基类不饱和化合物HPMA(2.60g,18.0mmol)以及四正丁基氯化铵(206mg,0.72mmol)置于聚合釜中,加入10mL甲苯溶解。将反应体系恒温至10℃,随后向体系中加入聚合催化剂三异丁基铝(7mL,7.7mmol),通过环氧乙烷钢瓶缓慢加入环氧乙烷(15.84g,360.0mmol),加料3小时随后在该温度下继续反应3小时。随后继续通过蠕动泵缓慢加入环氧单体TEOG(42.1g,180.0mmol),加料6小时随后在该温度下继续反应10小时。单体转化率由
1
接枝型聚醚大单体I-6的合成
该实施例中以丙烯酸羟乙酯(HEA)为环氧化合物单甲基五氧乙烯基缩水甘油醚(PEOG)开环聚合端羟基类不饱和化合物。
环氧单体PEOG聚合:于氮气气氛下,将干燥的开环聚合端羟基类不饱和化合物HEA(2.09g,18.0mmol)以及四正丁基氯化铵(206mg,0.72mmol)置于聚合釜中,加入10mL甲苯溶解。将反应体系恒温至10℃,随后向体系中加入聚合催化剂三异丁基铝(7mL,7.7mmol),通过蠕动泵缓慢加入环氧单体PEOG(28.9g,90.0mmol),加料6小时随后在该温度下继续反应7小时。单体转化率由
1
实施例1:
接枝型聚醚型聚羧酸减水剂PCAP-1的合成
该实施例以所合成接枝型聚醚大单体I-1、异戊烯基聚乙二醇单甲醚(TPEG,2400kg/mol)以及丙烯酸AA为共聚单体,通过普通自由基聚合方法合成接枝聚醚型聚羧酸减水剂PCAP-1。在该聚合体系中使用偶氮类引发剂偶氮二异丁脒盐酸盐V50,巯基丙酸BMA为链转移试剂。
取内置磁子的500mL三口瓶,向其中加入50mL去离子水以及异戊烯基聚乙二醇单甲醚(120g,30.0mmol,60wt%),通入氮气除氧10分钟后,升温至60℃。在搅拌状态下,滴加接枝型聚醚大单体I-1(37.23g,10.05mmol)、丙烯酸AA(1.158g,160.8mmol)、偶氮二异丁脒盐酸盐V50(1.095mg,4.05mmol,2mol%)、巯基丙酸BMA(315mg,4.05mmol,2mol%)及去离子水(90g)的混合溶液,滴加时长设置为4小时。滴加结束后,在该温度下继续反应2小时。聚合期间,每隔一小时从聚合体系取样,测定GPC以确定聚合体系单体转化率。聚合结束后,使用截留分子量为5000kg/mol的透析袋提纯聚合体系,以除去体系中残留单体,透析过程中每隔12小时置换透析用水,透析后使用旋转蒸发仪除水,并在真空条件下干燥。所合成接枝聚醚型聚羧酸减水剂的化学结构由傅里叶红外光谱仪Bruker Tensor-27 FT-IRspectrometer进行表征,如图1所示。Cov.=84%,M
实施例2:
接枝型聚醚型聚羧酸减水剂PCAP-2的合成
该实施例以所合成接枝型聚醚大单体I-3、异戊烯基聚乙二醇单甲醚(TPEG,2400kg/mol)以及丙烯酸AA为共聚单体,通过普通自由基聚合方法合成接枝聚醚型聚羧酸减水剂PCAP-2。在该聚合体系中使用偶氮类引发剂偶氮二异丁脒盐酸盐V50,巯基丙酸BMA为链转移试剂。
取内置磁子的500mL三口瓶,向其中加入50mL去离子水以及异戊烯基聚乙二醇单甲醚(120g,30.0mmol,60wt%),通入氮气除氧10分钟后,升温至60℃。在搅拌状态下,滴加接枝型聚醚大单体I-3(39.01g,10.05mmol)、丙烯酸AA(1.158g,160.8mmol)、偶氮二异丁脒盐酸盐V50(1.095mg,4.05mmol,2mol%)、巯基丙酸BMA(315mg,4.05mmol,2mol%)及去离子水(90g)的混合溶液,滴加时长设置为4小时。滴加结束后,在该温度下继续反应2小时。聚合期间,每隔一小时从聚合体系取样,测定GPC以确定聚合体系单体转化率。Cov.=96%,M
实施例3:
接枝型聚醚型聚羧酸减水剂PCAP-3的合成
该实施例以所合成接枝型聚醚大单体I-3、异戊烯基聚乙二醇单甲醚(TPEG,2400kg/mol)以及丙烯酸AA为共聚单体,通过普通自由基聚合方法合成接枝聚醚型聚羧酸减水剂PCAP-3。在该聚合体系中使用偶氮类引发剂偶氮二异丁脒盐酸盐V50,巯基丙酸BMA为链转移试剂。
取内置磁子的500mL三口瓶,向其中加入50mL去离子水以及异戊烯基聚乙二醇单甲醚(80g,20.0mmol,60wt%),通入氮气除氧10分钟后,升温至60℃。在搅拌状态下,滴加接枝型聚醚大单体I-3(78.38g,20.0mmol)、丙烯酸AA(1.158g,160.8mmol)、偶氮二异丁脒盐酸盐V50(1.095mg,4.05mmol,2mol%)、巯基丙酸BMA(315mg,4.05mmol,2mol%)及去离子水(90g)的混合溶液,滴加时长设置为4小时。滴加结束后,在该温度下继续反应2小时。聚合期间,每隔一小时从聚合体系取样,测定GPC以确定聚合体系单体转化率。Cov.=97%,M
对比例1:不使用本发明的接枝型聚醚大单体
聚羧酸减水剂PCA-1的合成
该实施例以异戊烯基聚乙二醇单甲醚(TPEG,2400kg/mol)以及丙烯酸AA为共聚单体,使用与接枝聚醚型聚羧酸减水剂合成相类似的合成方法合成常规聚羧酸减水剂PCA-1。
取内置磁子的500mL三口瓶,向其中加入50mL去离子水以及异戊烯基聚乙二醇单甲醚(160g,40.0mmol,60wt%),通入氮气除氧10分钟后,升温至60℃。在搅拌状态下,滴加丙烯酸AA(1.158g,160.8mmol)、偶氮二异丁脒盐酸盐V50(1.095mg,4.05mmol,2mol%)、巯基丙酸BMA(346mg,4.45mmol,2.2mol%)及去离子水(90g)的混合溶液,滴加时长设置为4小时。滴加结束后,在该温度下继续反应2小时。聚合期间,每隔一小时从聚合体系取样,测定GPC以确定聚合体系单体转化率。Cov.=95%,M
对比例2:不使用本发明的接枝型聚醚大单体
接枝型聚醚型聚羧酸减水剂PCA-2的合成
该实施例以异戊烯基聚乙二醇单甲醚(TPEG,2400kg/mol)以及丙烯酸AA为共聚单体,使用与接枝聚醚型聚羧酸减水剂合成相类似的合成方法合成常规聚羧酸减水剂PCA-2。
取内置磁子的500mL三口瓶,向其中加入50mL去离子水以及异戊烯基聚乙二醇单甲醚(160g,40.0mmol,60wt%),通入氮气除氧10分钟后,升温至60℃。在搅拌状态下,滴加丙烯酸AA(1.158g,160.8mmol)、偶氮二异丁脒盐酸盐V50(1.095mg,4.05mmol,2mol%)、巯基丙酸BMA(315mg,4.05mmol,2mol%)及去离子水(90g)的混合溶液,滴加时长设置为4小时。滴加结束后,在该温度下继续反应2小时。聚合期间,每隔一小时从聚合体系取样,测定GPC以确定聚合体系单体转化率。Cov.=94%,M
测试例1:水泥净浆流动度试验
本测试方法根据《GB/T8077-2012混凝土外加剂匀质性试验方法》中水泥净浆流动度测试方法进行。实验温度为25℃,水泥使用江南小野田水泥P·II 52.5级,减水剂掺量以水泥质量为基准,按折固量计算。在水泥净浆搅拌机中,分别加入本发明制备的接枝聚醚型聚羧酸减水剂PCAP-1、PCAP-2、PCAP-3以及对比样PCA-1、PCA-2水溶液,87g水以及300g鹤林P.O.42.5水泥,立即搅拌混合(慢速120s,停15s,快速120s)。将搅拌好的净浆注入截锥圆模内,提起截锥圆模,测定水泥净浆在玻璃平面上自由流淌的最大直径。分别在30分钟、60分钟和90分钟重复测试上述净浆流动度,比较其流动度的经时损失,并将数据记录于表1中。
表1
由表1实验结果可知,本发明所述接枝聚醚型聚羧酸减水剂PCAP-1、PCAP-2、PCAP-3与常规聚羧酸减水剂PCA-1、PCA-2相比具有明显优势,掺接枝聚醚型聚羧酸减水剂在初始减水率方面差别虽不明显,但其水泥净浆流动度经时损失相对更小。通过PCAP-2与PCAP-3的对比发现,随着接枝聚醚在聚羧酸减水剂中所占比例提高,聚合中单体转化率有所提高并净浆流动度增大,净浆流动度损失减小。推测接枝聚醚的引入,增大了吸附在水泥颗粒表面的聚羧酸减水剂空间位阻作用,阻碍了水泥颗粒表面的水化进程,因而接枝聚醚型聚羧酸减水剂在保坍性能方面表现更加优异。
测试例2:粘土适应性
通过净浆流动度实验评价所述接枝聚醚型聚羧酸减水剂在粘土适应性方面的应用效果。所采用方法与测试例1相类似,不同之处在于,测试例2使用与水泥等质量替换的方式掺入少量蒙脱土与高岭土的混合物,其中,蒙脱土和高岭土的质量比例为1:1。其初始净浆流动度及流动度经时损失记录于表2中。
表2
由表2实验结果可知,使用1wt%的粘土替换水泥后,掺接枝聚醚型聚羧酸减水剂的水泥净浆流动度降低幅度较小,对保坍性能的影响较小;此外,由PCAP-2与PCAP-3的结果对比发现,随着接枝聚醚型聚羧酸减水剂中接枝聚醚含量提高,其受粘土的影响效果降低。相比之下常规聚羧酸减水剂受粘土的影响十分显著,初始净浆流动度下降明显,且经时损失增大;对比表1和表2中的数据结果,可以看出增大聚羧酸折固掺量到0.15%后才能达到与不含粘土净浆流动度相近效果。因此,以上实验结果表明,接枝聚醚型聚羧酸减水剂相比常规聚羧酸减水剂在粘土适应性方面存在明显优势,在相同流动度条件下可有效降低聚羧酸减水剂所需掺量。
测试例3:接枝聚醚型聚羧酸减水剂在水泥颗粒及蒙脱土表面的吸附量
采用总有机碳法对接枝聚醚型聚羧酸减水剂在水泥颗粒及蒙脱土表面的吸附量进行比较。其测定方法如下:分别称取水泥或蒙脱土1g,加入50mL含1g/L接枝聚醚型聚羧酸减水剂PCAP-1、PCAP-2及PCAP-3的去离子水溶液,磁力搅拌10分钟,高速离心取上清液2mL并稀释20倍,使用总有机碳测试仪测定上清液中有机碳含量,以1g/L接枝聚醚型聚羧酸减水剂PCAP-1、PCAP-2及PCAP-3的去离子水溶液中总有机碳含量减去上清液中有机碳含量即为水泥颗粒或蒙脱土所吸附聚羧酸含量。其接枝聚醚型聚羧酸减水剂PCAP-1、PCAP-2及PCAP-3在水泥及蒙脱土颗粒表面的吸附数据如表3所示。
表3
由表3实验结果可知,接枝聚醚型聚羧酸减水剂PCAP-1、PCAP-2与PCAP-3受其紧密的侧链结构影响,其在水泥颗粒表面吸附量明显高于常规聚羧酸减水剂PCA-1、PCA-2,且由PCAP-2与PCAP-3吸附数据对比可知侧链结构中接枝聚醚含量越高水泥吸附量越高。常规聚羧酸减水剂PCA-1、PCA-2在蒙脱土表面的吸附量明显高于水泥颗粒表面吸附量,而接枝聚醚型聚羧酸减水剂PCAP-1、PCAP-2与PCAP-3在蒙脱土吸附量仅略高于其在水泥颗粒表面吸附量。因此,在粘土含量高的水泥中常规聚羧酸减水剂的减水保坍性能较差,而接枝聚醚型聚羧酸减水剂受粘土影响相对较小,仍能保持较好的水泥净浆流动度。
测试例4:不同石粉含量机制砂的适应性
本实施例通过砂浆实验评价本发明所述接枝聚醚型聚羧酸减水剂在不同石粉含量的机制砂情况下的适应性和性能稳定性。机制砂中石粉含量差别较大,这显著影响机制砂混凝土的工作性,也明显反映出减水剂适应性。水泥采用鹤林牌P.O.42.5水泥,机制砂采用水洗花岗岩机制砂,细度模数为2.8,亚加蓝值为0.6g/kg,掺加纯石粉和粘土石粉,后按照0%,10%,20%质量比配制成不同石粉含量的机制砂,研究机制砂的石粉含量对所述接枝聚醚型聚羧酸减水剂饱和掺量的影响,结果如表4所示。
表4
由表5中不同石粉含量机制砂砂浆流动度结果可知,随机制砂中石粉含量增加,减水剂饱和掺量逐步增大;对比接枝聚醚型聚羧酸减水剂PCAP-2、PCAP-3与常规聚羧酸减水剂PCA-2,PCAP-2与PCAP-3在机制砂中饱和点掺量更低,表现出更高的减水率和分散性;在石粉含量较高的机制砂中,掺PCAP-2与PCAP-3的砂浆流动度随减水剂掺量变化较小,而PCA-2的砂浆流动度随减水剂掺量变化较大,PCAP-2与PCAP-3表现出更低的减水剂敏感性。
尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。
- 一种枯烯基聚羧酸系减水剂聚醚大单体及其制备方法和应用
- 一种基于木质素基聚醚单体的聚羧酸减水剂及其制备方法和在混凝土中的应用