一种通过调节胞内PLP供给提高1,5-戊二胺从头合成能力的方法
文献发布时间:2024-04-18 19:58:26
技术领域
本发明涉及一种通过调节胞内PLP供给提高1,5-戊二胺从头合成能力的方法,属于生物技术领域。
背景技术
1,5-戊二胺是一种重要的化学品,可以作为原料合成一系列具有重要应用的高附加值产品,例如具有特殊性能的高档聚氨酯胶黏剂、聚氨酯弹性体,其应用覆盖聚氨酯、聚酯、涂料、染料、农业、医药等多个领域,具有广阔的市场前景。目前1,5-戊二胺从头合成效率低下限制了其产能和应用,但全球市场每年消耗约3000万t聚酰胺产品,而且市场需求量程呈逐年上涨的趋势。
目前全球工业上1,5-戊二胺大部分以不可再生的石油化石原料通过化学法以及全细胞催化的生物转化法合成,其生产装置较为复杂,生产费用较高,同时还会造成一定程度上的环境污染。不仅如此,工业生产设备通常面临设备装置要求高、设备腐蚀问题严重、催化剂稳定性差等问题。
利用从头发酵的方法合成1,5-戊二胺可以减少化石能源的消耗以及节约成本,提高循环经济水平,实现低碳生产。目前,发酵法合成1,5-戊二胺的研究主要集中在使用葡萄糖、木质纤维素等生物质经过发酵法进行合成,但是由于漫长的代谢路径、1,5-戊二胺的毒性以及关键合成酶赖氨酸脱羧酶的辅因子短缺等问题一直尚未得到良好的解决,导致从头发酵的1,5-戊二胺产能一直处于较低水平,无法进行工业化生产。目前,采用从头发酵生产1,5-戊二胺研究最多的是南京工业大学陈可泉团队,并且也是目前通过从头发酵生产1,5-戊二胺产量最高(58.7g/L)的团队。因此,开发一种清洁高效的1,5-戊二胺微生物从头合成法很有必要。
通过从头发酵的方法生产1,5-戊二胺,不仅反应温和,而且生产成本低。因此,开发基于赖氨酸为前体生物生产1,5-戊二胺的生物制造工业对于环境保护及提高循环经济水平都具有重大意义。
发明内容
为解决上述问题,本发明通过协同两种PLP强化途径,并对该强化途径中的基因进行筛选,得到最优组合为MypdxT和ScpdxS,并进一步对启动子表达元件以及诱导时间进行优化,最终得到了一种高效、高产,在温和条件下从头发酵生产1,5-戊二胺的新方法。
本发明的第一个目的是提供一种提高重组大肠杆菌从头合成1,5-戊二胺产量的方法,所述方法是在大肠杆菌宿主中过表达SEQ ID NO.1所示的赖氨酸脱羧酶基因(cadA)、SEQ ID NO.2所示的4-磷酸羟基-L-苏氨酸脱氢酶基因(pdxA)、SEQ ID NO.9所示的谷氨酰胺酶亚基基因(MypdxT)、SEQ ID NO.12所示的PLP合酶亚基基因(ScpdxS)。
进一步地,采用启动子P
进一步地,采用启动子P
进一步地,采用第一启动子启动4-磷酸羟基-L-苏氨酸脱氢酶基因的表达,采用第二启动子启动谷氨酰胺酶亚基基因和PLP合酶亚基基因的表达,其中,所述第一启动子选自P
进一步地,启动子P
进一步地,所述方法为采用重组大肠杆菌进行发酵生产,发酵生产时包括采用以下步骤添加诱导剂:菌株接种至发酵培养基的第0~3小时加入诱导剂。
进一步地,发酵的初始葡萄糖浓度为28-32g/L。
进一步地,发酵条件为:36-38℃,170-220rpm,pH控制在6.6-6.7,残糖控制在0-10g/L,氨氮含量控制在0.15%-0.18%。
进一步地,诱导剂的浓度为0.3-0.6mmol/L。
进一步地,发酵培养基包括以下组分:无水葡萄糖28-32g/L,七水硫酸镁0.2-0.6g/L,玉米浆蛋白干粉8-12g/L,硫酸铵1.5-2.0g/L,磷酸二氢钾0.4-0.8g/L,磷酸氢二钾1.2-1.6g/L。
进一步地,以E.coli MG1655为出发菌株。
进一步地,以pEM为表达载体。
本发明的第二个目的是提供一种大肠杆菌重组菌,所述大肠杆菌重组菌是在大肠杆菌宿主中过表达SEQ ID NO.1所示的赖氨酸脱羧酶基因(cadA)、SEQ ID NO.2所示的4-磷酸羟基-L-苏氨酸脱氢酶基因(pdxA)、SEQ ID NO.9所示的谷氨酰胺酶亚基基因(MypdxT)、SEQ ID NO.12所示的PLP合酶亚基基因(ScpdxS)。
进一步地,采用启动子P
进一步地,采用启动子P
进一步地,采用第一启动子启动4-磷酸羟基-L-苏氨酸脱氢酶基因的表达,采用第二启动子启动谷氨酰胺酶亚基基因和PLP合酶亚基基因的表达,其中,所述第一启动子选自P
本发明的第三个目的是提供上述大肠杆菌重组菌的构建方法,包括以下步骤:
S1、将基因片段cadA与pdxA连接并插入到载体pEM上,得到重组质粒pEM-cadA-pdxA;
S2、将基因片段cadA、MypdxT与ScpdxS连接并插入到载体pEM上,得到重组质粒pEM-cadA-pdxS/T;
S3、将重组质粒pEM-cadA-pdxA和pEM-cadA-pdxS/T转化入大肠杆菌宿主中,构建得到所述大肠杆菌重组菌。
进一步地,还包括将pdxA和pdxS/T的原启动子替换为上述第一启动子和第二启动子的步骤。
本发明的第四个目的是提供上述大肠杆菌重组菌在制备含1,5-戊二胺或其衍生物的产品中的应用。
本发明的有益效果:
本发明提供了一种发酵法从头生产1,5戊二胺的方法,其能够在温和条件下从葡萄糖生产1,5-戊二胺。该反应成本更低,条件更加温和,以葡萄糖为底物,加入重组大肠杆菌,摇瓶发酵48h后生产1,5-戊二胺的浓度为11.23g/L。
附图说明
图1为发酵过程中的生物量。
图2为发酵过程中的1,5-戊二胺积累量。
图3为发酵过程中的赖氨酸积累量。
图4为不同强度启动子操纵PLP胞内供给的1,5-戊二胺积累量。
图5为不同诱导时间下的1,5-戊二胺积累量。
具体实施方式
下面结合附图和具体实施例对本发明作进一步说明,以使本领域的技术人员可以更好地理解本发明并能予以实施,但所举实施例不作为对本发明的限定。
本发明涉及的方案如下:
大肠杆菌本身无法通过发酵法高效合成1,5-戊二胺,为此在大肠杆菌中引入来自Escherichia coli的赖氨酸脱羧酶基因cadA、来自Escherichia coli的4-磷酸羟基-L-苏氨酸脱氢酶基因pdxA、来自M.tuberculosis的谷氨酰胺酶亚基基因pdxT以及来自S.cerevisiae的PLP合酶亚基基因pdxS获得了一种高效生产1,5-戊二胺重组大肠杆菌(lysWT-3)。随后对重组大肠杆菌lysWT-3进行表达元件(具体地,首先采用原启动子P
本发明涉及的材料如下:
LB培养基(g/L):酵母提取物5,胰化蛋白胨10,氯化钠10,NaOH调pH至7.0,121℃、20min高压灭菌。
TB培养基(g/L):胰蛋白胨11.8,酵母提取物23.6,K2HPO4 9.4,g/L KH2PO4 2.2,甘油4ml/L,pH至7.0,121℃、20min高压灭菌。
斜面培养基(g/L):胰蛋白胨(Oxoid)10,氯化钠5,丙酮酸钠0.5,酵母抽提物(Oxoid)5,琼脂粉20。每只试管装液量20~25mL,试管塞为棉塞,121℃灭菌15min。
一级种子培养基(g/L):酵母抽提物5,蔗糖3,胰蛋白胨3,磷酸二氢钾0.5,磷酸氢二钾1.0,pH=7.5~7.6,121℃灭菌15min。
二级种子培养基(g/L):无水葡萄糖20,玉米浆蛋白干粉5,磷酸二氢钾1.44,硫酸铵14.4,pH=7.5~7.6,121℃灭菌20min,葡萄糖115℃灭菌15min。发酵培养基(g/L):无水葡萄糖30,七水硫酸镁0.4,玉米浆蛋白干粉10.36,硫酸铵1.8,磷酸二氢钾0.6,磷酸氢二钾1.4,121℃灭菌15min,葡萄糖115℃灭菌15min。
实施例1重组大肠杆菌lysWT-1和lysWT-2的构建
1、重组质粒pEM-cadA的构建
(1)以cadA U和cadAD为引物,以cadA基因(核酸序列如SEQ ID NO.1所示)为模板,扩增获得带酶切位点的cadA片段。
cadA U:
GGATCGCATCACCATCACCATCACGGATCCATGAACGTTATTGCAAT ATTGAATCAC;
cadA D:
AGATCTACCAGACTCGAGTTATTTTTTGCTTTCTTCTTTCAATACCT;
(2)用pEM U和pEM D反向扩增获得线性化载体pEM。
pEM U:CTCGAGTCTGGTAGATCT;
pEM D:GGATCCGTGATGGTGATG;
(3)将步骤(2)所得线性化载体与步骤(1)所得基因片段cadA连接,获得重组质粒pEM-cadA。
2、重组质粒pEM-cadA-pdxA的构建
(1)以pdxA U和pdxA D为引物,以人工合成密码子优化后的pdxA基因(核酸序列如SEQ ID NO.2所示)为模板扩增获得pdxA片段。
pdxA U:
AGAAAGCAAAAAATAACTCGAGTCTGGTAGATCTGGATCCTTGACA ATTAATCATCGGCTCGTATAATGTGTGGAATTGTGAGCGGATAACAATTTC ACACAGGAAACAGAATTCATGGTTAAAACCCAACGTGTT;
pdxA D:
AGCCATATGGGTGGCAGCAGTCATTGGGTGTTAACAATCATTTTG;
(2)以pEM-cadA U和pEM-cadA D反向扩增获得线性化载体pEM-cadA。
pEM-cadA U:CTGCTGCCACCCATATG;
pEM-cadA D:GAATTCTGTTTCCTGTGTGAAAT;
(3)步骤(2)所得酶切质粒与步骤(1)所得基因片段pdxA连接,获得重组质粒pEM-cadA-pdxA。
3、重组大肠杆菌lysWT-1和lysWT-2的构建
(1)将重组质粒pEM-cadA导入大肠杆菌Escherichia coli MG1655的感受态中,在包含100mg/L氨苄霉素的LB平板上获得正确的重组菌株,命名为lysWT-1。
(2)将重组质粒pEM-cadA-pdxA导入大肠杆菌Escherichia coli MG1655的感受态中,在包含100mg/L氨苄霉素的LB平板上获得正确的重组菌株,命名为lysWT-2。
实施例2pdxS/T的筛选
1、重组质粒pET28a-StpdxT、pET28a-BlglsA、pET28a-PfpdxT、pET28a-GepdxT、pET28a-BcpdxT、pET28a-ScpdxT、pET28a-MypdxT、pET28a-GepdxS、pET28a-BcpdxS、pET28a-ScpdxS、pET28a-MypdxS、pET28a-PfpdxS的构建
(1)以StpdxT U和StpdxT D为引物,以人工合成密码子优化后的StpdxT基因(核酸序列如SEQ ID NO.3所示)为模板扩增获得StpdxT片段。
StpdxT U:
AGCAAATGGGTCGCGGATCCATGACAATAATGTCAAGTCAATTTCAGC;
StpdxT D:
GTGGTGGTGGTGGTGCTCGAGTTAGAAAACGCTCAGGCCAG;
(2)以BlglsA U和BlglsA D为引物,以人工合成密码子优化后的BlglsA基因(核酸序列如SEQ ID NO.4所示)为模板扩增获得BlglsA片段。
BlglsA U:
CAGCAAATGGGTCGCGGATCCTTAAATTGTAGGCACAACGAAGAAC;
BlglsA D:
GTGGTGGTGGTGGTGCTCGAGTTAAAAGATGGAAAGGCTATAATTCGCC;
(3)以PfpdxT U和PfpdxT D为引物,以人工合成密码子优化后的PfpdxT基因(核酸序列如SEQ ID NO.5所示)为模板扩增获得PfpdxT片段。
PfpdxT U:
AGCAAATGGGTCGCGGATCCATGAGTGAAATAACAATTGGAGTACTATCATT;
PfpdxT D:
GTGGTGGTGGTGGTGCTCGAGTTAAGAGTATTTGTAGTTCTTAACCTTCT CG;
(4)以GepdxT U和GepdxT D为引物,以人工合成密码子优化后的GepdxT基因(核酸序列如SEQ ID NO.6所示)为模板扩增获得GepdxT片段。
GepdxT U:
GTGGTGGTGGTGGTGCTCGAGTTAAGAGTATTTGTAGTTCTTAACCT TCT CG;
GepdxT D:
GTGGTGGTGGTGGTGCTCGAGTTACTTCAGGCTGCTGGTCA;
(5)以BcpdxT U和BcpdxT D为引物,以BcpdxT基因(核酸序列如SEQ ID NO.7所示)为模板扩增获得BcpdxT片段。
BcpdxT U:
AGCAAATGGGTCGCGGATCCATGTTAACAATAGGTGTACTAGGACT;BcpdxT D:
TGGTGGTGGTGGTGCTCGAGTTATACAAGTGCCTTTTGCTTATATTC CT;
(6)以ScpdxT U和ScpdxT D为引物,以ScpdxT基因(核酸序列如SEQ ID NO.8所示)为模板扩增获得ScpdxT片段。
ScpdxT U:
CAGCAAATGGGTCGCGGATCCATGCACAAAACCCACAGTAC;
ScpdxT D:
TGGTGGTGGTGGTGGTGCTCGAGTTAATTAGAAACAAACTGTCTGA TAA ACCAAT;
(7)以MypdxT U和MypdxT D为引物,以人工合成密码子优化后的MypdxT基因(核酸序列如SEQ ID NO.9所示)为模板扩增获得MypdxT片段。
MypdxT U:
AGCAAATGGGTCGCGGATCCGTAAGTGTACCCAGGGTCG;
MypdxT D:
GTGGTGGTGGTGGTGCTCGAGTTACGCGGCCGAGGT;
(8)以GepdxS U和GepdxS D为引物,以人工合成密码子优化后的GepdxS基因(核酸序列如SEQ ID NO.10所示)为模板扩增获得GepdxS片段。
GepdxS U:
CAGCAAATGGGTCGCGGATCCTTAGCTCTAACTGGAACAGATAGGG;
GepdxS D:
GTGGTGGTGGTGGTGCTCGAGTTACCAACCACGTTCTTGCATA;
(9)以BcpdxS U和BcpdxS D为引物,以BcpdxS基因(核酸序列如SEQ ID NO.11所示)为模板扩增获得BcpdxS片段。
BcpdxS U:
AGCAAATGGGTCGCGGATCCATGGCTCAAACAGGTACTGA;
BcpdxS D:
TGGTGGTGGTGGTGCTCGAGTTACCAGCCGCGTTCTTG;
(10)以ScpdxS U和ScpdxS D为引物,以ScpdxS基因(核酸序列如SEQ ID NO.12所示)为模板扩增获得ScpdxS片段。
ScpdxS U:
CAGCAAATGGGTCGCGGATCCATGACTGGAGAAGACTTTAAGATCA;
ScpdxS D:
GGTGGTGGTGGTGGTGCTCGAGTCACCACCCAATTTCGGAAAG;
(11)以MypdxS U和MypdxS D为引物,以人工合成密码子优化后的MypdxS基因(核酸序列如SEQ ID NO.13所示)为模板扩增获得MypdxS片段。MypdxS U:AGCAAATGGGTCGCGGATCCATGGACCCCGCTGGAAA;MypdxS D:TGGTGGTGGTGGTGCTCGAGTTACCAGCCACGCTGGG;
(12)以PfpdxS U和PfpdxS D为引物,以人工合成密码子优化后的PfpdxS基因(核酸序列如SEQ ID NO.14所示)为模板扩增获得PfpdxS片段。
PfpdxS U:
ACAGCAAATGGGTCGCGGATCCATGGAAAATCACAAAGATGACGC;PfpdxS D:
TGGTGGTGGTGGTGCTCGAGTTATTGCGGAGTCAGGAACTTG;
(13)以pET28a U和pET28a D反向扩增获得线性化载体pET28a。
pET28a U:CTCGAGCACCACCACC;
pET28a D:GGATCCGCGACCCATT;
(14)将步骤(13)所得线性化载体与步骤(1)~(12)所得基因片段StpdxT、BlglsA、PfpdxT、GepdxT、BcpdxT、ScpdxT、MypdxT、GepdxS、BcpdxS、ScpdxS、MypdxS、PfpdxS相连接,获得相应的重组质粒pET28a-StpdxT、pET28a-BlglsA、pET28a-PfpdxT、pET28a-GepdxT、pET28a-BcpdxT、pET28a-ScpdxT、pET28a-MypdxT、pET28a-GepdxS、pET28a-BcpdxS、pET28a-ScpdxS、pET28a-MypdxS、pET28a-PfpdxS。
(15)将步骤(14)所得的重组质粒pET28a-StpdxT、pET28a-BlglsA、pET28a-PfpdxT、pET28a-GepdxT、pET28a-BcpdxT、pET28a-ScpdxT、pET28a-MypdxT、pET28a-GepdxS、pET28a-BcpdxS、pET28a-ScpdxS、pET28a-MypdxS、pET28a-PfpdxS导入E.coli BL21(DE3)感受态中,构建重组菌株E.coli BL21(DE3)pET28a-StpdxT/pET28a-BlglsA/pET28a-PfpdxT/pET28a-GepdxT/pET28a-BcpdxT/pET28a-ScpdxT/pET28a-MypdxT/pET28a-GepdxS/pET28a-BcpdxS/pET28a-ScpdxS/pET28a-MypdxS/pET28a-PfpdxS。
2、收菌、获得纯酶
(1)首先,将菌株E.coli BL21(DE3)pET28a-StpdxT/pET28a-BlglsA/pET28a-PfpdxT/pET28a-GepdxT/pET28a-BcpdxT/pET28a-ScpdxT/pET28a-Mypd xT/pET28a-GepdxS/pET28a-BcpdxS/pET28a-ScpdxS/pET28a-MypdxS/pET28a-Pf pdxS分别以1%的接种量接种至液体LB培养基中,37℃,200rpm,培养24h。其次,再以5%的接种量转接至TB培养基中,37℃,200rpm,培养至OD
(2)将步骤(1)获得经诱导表达后收集的菌体,用100mM的PBS溶液(pH 7.4)重复洗涤三次后,在4℃、7000rpm条件下离心约15min后收集菌体;称取1g菌体,加入约10mL的结合液A(20mM磷酸钠、0.5mM NaCl、20mM咪唑、1%甘油,盐酸调pH至7.4),重悬菌体后,超声破碎10min左右;破碎液在4℃、7000rpm条件下离心10min左右,上清液经0.22um微孔滤膜过滤后,获得粗酶液。
(3)用结合液A(需10倍左右柱体积)平衡镍离子亲和层析柱;将上述制得的粗酶液倒入柱子至液体完全流出;先用结合液A冲洗柱子以除去杂蛋白,再加入洗脱液C(20mM磷酸钠、0.5mM NaCl,50mM咪唑,pH 7.4)清洗柱子至液体流出,最后加入洗脱液B(20mM磷酸钠、0.5mM NaCl、500mM咪唑,pH 7.4)收集目的蛋白。
3、酶活和酶动力学参数的测定
(1)谷氨酰胺酶活以及酶动力学参数的测定—偶联酶测定
通过与谷氨酸脱氢酶(NCBI Gene ID:946802)的偶联反应测定PdxT活性。在该测定中,谷氨酰胺被PdxT水解为谷氨酸,谷氨酸通过谷氨酸脱氢酶转化为α-酮戊二酸,同时还原3-乙酰吡啶腺嘌呤二核苷酸(APAD),NAD的类似物,可改变谷氨酸脱氢酶反应的不利平衡。它的减少可以观察到363nm处吸光度的增加。样品在37℃下测定,总反应体积为300μL,含有50mM Tris-Cl缓冲液(pH 8.0),0.5mM APAD,7单位谷氨酸脱氢酶和10mM谷氨酰胺。为了测定动力学常数,使用了4μM PdxT。使用采用牛血清白蛋白作为标准品的Bradford试剂(Pierce)测定蛋白质浓度。
(2)PLP酶活和酶动力学参数的测定
通过紫外-可见分光光度法监测PdxS的活性,其中在414nm处出现最大值表明希夫碱的酶促形成,可能是由生成的反应产物和测定中使用的Tris缓冲液(2-氨基-2-羟甲基-1,3-丙二醇)的伯胺形成的。反应在50mM Tris-Cl缓冲液(pH 8.0),37℃下进行,含有40μM分离的PdxS以及0.5mM底物:核糖-5-磷酸和DL-甘油醛3-磷酸(1mM)。作为氮源,加入10mM谷氨酰胺或10mM硫酸铵。
4、结果
表1不同来源的谷氨酰胺酶的动力学参数
表2不同来源的PLP合酶的动力学参数
因此,选用ScpdxS和MypdxT进行后续实验。
实施例3重组大肠杆菌lysWT-3的构建
1、重组质粒pEM-cadA-pdxS/T的构建
(1)以ScpdxS U2和ScpdxS D2为引物,以ScpdxS基因(核酸序列如SEQ ID NO.12所示)为模板扩增获得ScpdxS片段。
ScpdxS U2:
AGTCTGGTAGATCTGGATCCTTGACAATTAATCATCGGCTCGTATAAT
GTG
TGGAATTGTGAGCGGATAACAATTTCACACAGGAAACAGAATTCATGACTGGAGAAGACTTTAAGATCA;
ScpdxS D2:
GCTACCGCTACCGCTACCCCACCCAATTTCGGAAAGTCTTA;
(2)以MypdxT U2和MypdxT D2为引物,以人工合成密码子优化后的MypdxT基因(核酸序列如SEQ ID NO.9所示)为模板扩增获得MypdxT片段。
MypdxT U2:
GGTAGCGGTAGCGGTAGCGGATCCGTAAGTGTACCCAG;
MypdxT D2:
GGCAGCAGGTGGTGCTCGAGGTGGTGCTCGAGTTACG;
(3)以pEM-cadA U2和pEM-cadA D2反向扩增获得线性化载体pEM-cadA。
pEM-cadA U2:CACCACCTGCTGCCAC;
pEM-cadA D2:AGCAAATAAATTTTTTATGACATATGGGTGGCAGCAG;
(4)将步骤(3)所得线性化载体与步骤(1)、(2)所得基因片段pdxS和pdxT连接,获得重组质粒pEM-cadA-pdxS/T。
2、重组菌株的构建
将重组质粒pEM-cadA-pdxA及pEM-cadA-pdxS/T转化入大肠杆菌Escherichiacoli MG1655的感受态中,在包含100mg/L氨苄霉素的LB平板上获得正确的重组菌株,命名为lysWT-3。
实施例4发酵法合成1,5-戊二胺
首先,选取大肠杆菌lysWT-1、lysWT2、lysWT-3,在斜面固体培养基上活化24h;
其次,将活化后的大肠杆菌lysWT-1、lysWT2、lysWT-3接种于一级种子培养基中,37℃扩大培养7.5-8h;然后,将活化的一级种子培养基以10%的接种量转接到二级种子培养基中,在37℃下扩大培养14-16h;再然后,将活化的二级种子培养基以15%-16%的接种量转接到发酵培养基中,在37℃下培养;
最后,在培养初始阶段,添加终浓度为0.42mmol/L的IPTG,诱导表达赖氨酸脱羧酶,催化赖氨酸合成1,5-戊二胺,所述发酵条件为:37±1℃,190rpm,pH控制在6.6-6.7,残糖控制在0-10g/L,氨氮含量控制在0.15%-0.18%,发酵周期为48h。
发酵结束后,在4℃,12000r/min的条件下离心10min,收集上清液,利用高效液相色谱检测1,5-戊二胺以及赖氨酸的产生情况,检测条件:首先用DEEMM衍生L-赖氨酸和1,5-戊二胺,衍生化体系包括:75μL反应混合物样品、4.5μL DEEMM、70.5μL蒸馏水、150μL 100%甲醇和450μL 50mmol/L硼酸盐缓冲液(pH 9.0)。将衍生化后混合物在涡旋混合仪中剧烈混合,然后在70℃加热2h以去除多余的DEEMM和副产物。随后,将样品以10000r/min离心5min后,用0.22μm膜过滤,然后用HPLC系统(SB-C18色谱柱)进行分析。柱温度设定在35℃,进样量10μL,流动相由100%乙腈(A)和25mmol/L乙酸钠水溶液(pH 4.8)(B)组成,流速1mL/min,溶剂梯度如下:0~2min,20%~25%A;2~20min,25%~60%A;20~25min,60%~20%A。根据标准曲线计算即得各组分实际浓度。
结果发现:(1)lysWT-1、lysWT-2、lysWT-3菌株的最大OD
实施例5强化PLP胞内供给-优化启动子
本实施例中从头合成1,5-戊二胺的具体方法与实施例4相同,区别仅在于,改变启动子,使两个PLP合成模块的启动子分别为P
实施例6发酵条件-摇瓶水平优化诱导时间
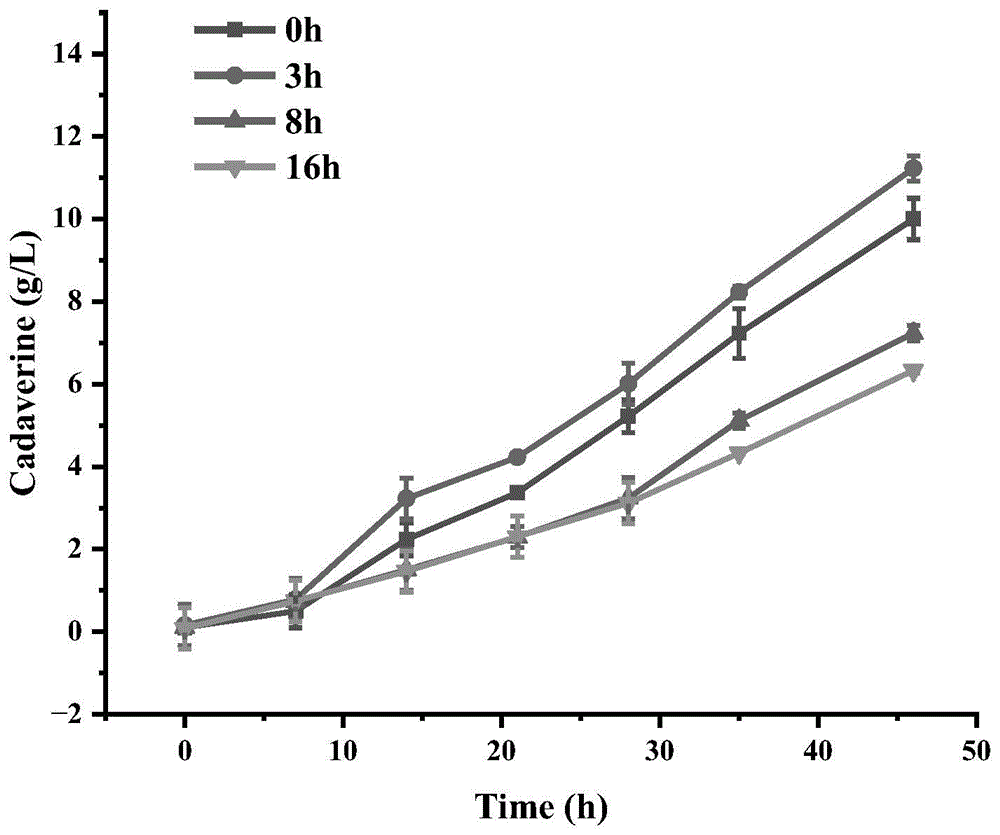
本实施例中从头合成1,5-戊二胺的具体方法与实施例5中的LysWT-5+A组相同,区别仅在于,改变加入IPTG的时间,使诱导起始时间分别改变为发酵第0h、3h、8h、16h,其他反应条件不变,48h后反应液中1,5-戊二胺的浓度分别为10.00g/L、11.23g/L、7.23g/L、6.33g/L(图5)。
显然,上述实施例仅仅是为清楚地说明所作的举例,并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引申出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
- 生产1,5-戊二胺的方法
- 1,5-戊二胺的制造方法、1,5-戊二胺、1,5-戊二异氰酸酯、1,5-戊二异氰酸酯的制造方法、聚异氰酸酯组合物、及聚氨酯树脂
- 一种1,5-戊二胺的提取方法及其所得的1,5-戊二胺产品