有机发光化合物及包含其的有机电致发光器件
文献发布时间:2023-06-19 10:13:22
技术领域
本发明涉及5,6-取代的三嗪基喹啉衍生物有机发光化合物及包含其的有机电致发光器件。
背景技术
电致发光器件(electroluminescence device,EL device)为自发光显示器件,具有响应速度快且视角宽的优点。1987年,伊士曼柯达公司(Eastman Kodak)公司首次研发了将低分子芳香二胺和铝配合物用作发光层材料的有机电致发光器件[Appl.Phys.Lett.51,913,1987]。
在有机电致发光器件中确定发光效率的最重要因素为发光材料,发光材料中的磷光材料可在理论上将发光效率改善至荧光材料的4倍为止。目前为止铱(III)配合物类和咔唑累的材料作为磷光发光材料周知,最近,在研究心得磷光材料。
有机电致发光现象的原理如下,即,当有机薄膜层介于阴极与阳极之间时,若向两个电极之间施加电压,则电子和空穴分别从阴极和阳极向有机薄膜层注入。向有机薄膜层注入的电子和空穴再次结合来形成激子(exciton),该激子再次回落到基态并发光。利用这种原理的有机电致发光器件通常由阴极和阳极及位于其之间的有机薄膜层,例如,包括空穴注入层、空穴传输层、发光层、电子传输层的有机薄膜层构成。
在有机电致发光器件中使用的材料大部分为纯有机物或由有机物和金属形成配合物的配位化合物,可根据用途分为空穴注入材料、空穴传输材料、发光材料、电子传输材料、电子注入材料等。其中,作为空穴注入材料或空穴传输材料,主要使用具有p型性质的有机材料,即,容易氧化且氧化时具有电化学稳定状态的有机物。另一方面,作为电子注入材料或电子传输材料,主要使用具有n型性质的有机材料,即,容易还原且在还原时具有电化学稳定状态的有机物。作为发光层材料,优选地,使用同时具有p型性质和n型性质的材料,即,在氧化和还原状态下均具有稳定形态的材料,优选地,为当形成激子时,将其转换为光的发光效率高的材料。因此,在本技术领域中需要研发具有如上所述的条件的新的有机材料。
发明内容
发明所要解决的问题
本发明的一实例提供具有适当的能级、电化学稳定性及热稳定性的5,6-取代的三嗪基喹啉衍生物有机发光化合物。
用于解决问题的方案
在本发明的一实例中,提供由下述化学式a表示的5,6-取代的三嗪基喹啉衍生物有机发光化合物。
化学式a:
在上述化学式a中,
L为单键或者选自由取代或未取代的C6~C30的亚芳基、取代或未取代的C10~C30的缩合亚芳基、取代或未取代的C5~C30的包含N、O、S及Si的杂亚芳基及它们的组合组成的组中的一种,上述L被取代的情况下的取代基选自由氢、氘、C1~C40的烷基、C1~C40的烷氧基、C6~C40的芳基、C5~C40的芳氧基、C5~C40的杂芳基及它们的组合组成的组中,
R为氢、氘、氰基、腈基、卤素基、羟基、C1~C40的烷基、C1~C40的烷氧基、取代或未取代的C6~C40的芳基、C5~C40的芳氧基、取代或未取代的C3~C40的包含N、O、S及Si的杂芳基、C6~C40的芳烷基或C3~C40的环烷基,上述Ar
N为0至5,
Ar
Ar
具体地,上述Ar
具体地,上述Ar
具体地,上述L可以为选自由下述组成的组中的一种化合物。
上述有机发光化合物可用作有机电致发光器件用材料中的发光层、空穴阻挡层、电子传输层或电子注入层物质。
在本发明的再一实例中,提供至少一个有机薄膜层夹在阴极与阳极之间的有机电致发光器件,上述有机薄膜层为包括至少一个发光层及电子传输区域的多层结构,上述有机薄膜层中的至少一个层单独包含上述5,6-取代的三嗪基喹啉衍生物有机化合物或包含2种以上的混合物。
上述电子传输区域介于阴极与发光层之间,包括电子注入层、电子传输层、同时具有电子注入功能及电子传输功能的功能层、缓冲层及空穴阻挡层中的至少一个层。
发明效果
上述5,6-取代的三嗪基喹啉衍生物有机发光化合物可优秀地满足可在有机电致发光器件中使用的物质所需的条件,例如,适当的能级、电化学稳定性及热稳定性等,可根据取代基起到有机电致发光器件所需的多种作用。
附图说明
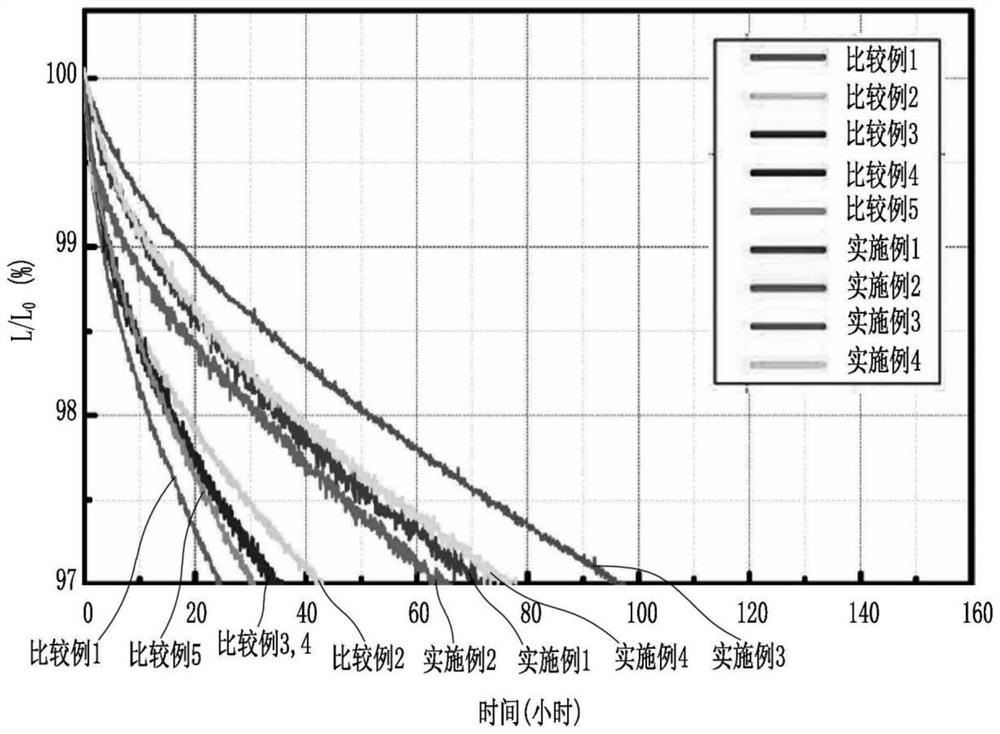
图1图2为示出在实施例1~9及比较例1~5中制备的有机电致发光器件的测定结果的寿命特性评价曲线图。
图3为示出在实施例10~13及比较例1、2及6中制备的有机电致发光器件的测定结果的寿命特性评价曲线图。
具体实施方式
以下,详细说明本发明的实例。但是,这仅为例示,本发明并不局限于此,本发明通过发明要求保护范围来定义。
在本说明书中,除非另行定义,则被“取代”的情况包括被选自由C1-C12的烷基、氨基、腈基、C3-C7的环烷基、C2-C12的烯基、C3-C7的环烯基、C2-C50的炔基、C5-C50的环炔基、氰基,C1-C12的烷氧基、C6-C60的芳基及C7-C60的芳烷基及它们的组合组成的组中的取代基取代的情况。
在本说明书中,除非另行定义,则“它们的组合”意味着两个以上的取代基通过连接基键合或两个以上的取代基缩合并键合。
在本说明书中,除非另行定义,则“杂”意味着在一个化合物或取代基中包含杂原子,上述杂原子可为选自由N、O、S、P及它们的组合组成的组中一个。例如,可意味着在上述一个化合物或取代基中包含1个至3个杂原子且其余为碳的情况。
在本发明的一实例中,提供由新的下述化学式a表示的5,6-取代的三嗪基喹啉衍生物有机发光化合物。
化学式a:
在上述化学式a中,
L为单键或者选自由取代或未取代的C6~C30的亚芳基、取代或未取代的C10~C30的缩合亚芳基、取代或未取代的C5~C30的包含N、O、S及Si的杂亚芳基及它们的组合组成的组中的一种,上述L被取代的情况下的取代基选自由氢、氘、C1~C40的烷基、C1~C40的烷氧基、C6~C40的芳基、C5~C40的芳氧基、C5~C40的杂芳基及它们的组合组成的组中,
R为氢、氘、氰基、腈基、卤素基、羟基、C1~C40的烷基、C1~C40的烷氧基、取代或未取代的C6~C40的芳基、C5~C40的芳氧基、取代或未取代的C3~C40的包含N、O、S及Si的杂芳基、C6~C40的芳烷基或C3~C40的环烷基,上述Ar
N为0至5,
Ar
Ar
具体地,上述Ar
具体地,上述Ar
具体地,上述L可以为选自由下述组成的组中的一种化合物。
例如,上述5,6-取代的三嗪基喹啉衍生物有机发光化合物可以为在下述表1中记载的1至324中的一种化合物。
表1
在将上述5,6-取代的三嗪基喹啉衍生物有机发光化合物用作有机电致发光器件用材料的情况下,可优秀地满足可在有机电致发光器件中使用的物质所需的所有条件,例如,适当的能级、电化学稳定性及热稳定性等,可根据取代基起到有机电致发光器件所需的各种作用。
在本发明的另一实例中,在至少一个有机薄膜层夹在阴极与阳极之间的有机电致发光器件中,上述有机薄膜层单独包含上述5,6-取代的三嗪基喹啉衍生物有机发光化合物或包含2种以上的混合物。
更具体地,提供如下的有机电致发光器件,即,上述有机薄膜层为包括至少一个发光层及电子传输区域的多层结构,上述有机薄膜层中的至少一个层单独包含上述5,6-取代的三嗪基喹啉衍生物有机发光化合物或包含2种以上的混合物。
包含于上述有机电致发光器件的有机薄膜层的上述5,6-取代的三嗪基喹啉衍生物有机发光化合物为由上述化学式a表示的化合物,与之有关的详细说明如前所述。
上述电子传输区域介于阴极与发光层之间,可包括选自由电子注入层、电子传输层、同时具有电子注入功能及电子传输功能的功能层、缓冲层及空穴阻挡层中的至少一个层形成的组中的至少一个层,可根据所需的用途适当的选择其。
上述空穴注入层、空穴传输层、功能层、缓冲层、电子屏蔽层、发光层、空穴阻挡层、电子传输层、电子注入层等分别可使用公知的物质来形成,或者,它们的至少一个可包含一种以上的由上述化学式a表示的5,6-取代的三嗪基喹啉衍生物有机发光化合物。
与包含于上述有机薄膜层的由上述化学式a表示的5,6-取代的三嗪基喹啉衍生物有机发光化合物有关的详细说明如前所述。
以下,记载本发明的实施例及比较例。下述实施例仅为本发明的一实施例,本发明并不限定于下述实施例。
实施例
以下,具体例示反应例及比较例,本发明并不限定于下述反应例及实施例。在以下反应例中,以向最终生成物的编号赋予序列号的方式标记中间体化合物。例如,由化合物[1]标记化合物1,由[1-1]等标记上述化合物的中间体化合物。在本说明书中,由在上述表1记载的化合物编号标记化合物的编号。例如,由“化合物1”标记在表1中由“1”表示的化合物。
合成例1:制备化合物[1]
反应式1:
制备中间体化合物[1-1]
在氮气氛下,向2L的反应烧瓶投入20g(125.6mmol)的6-甲氧基喹啉和300ml的二氯甲烷并搅拌。混合7.1ml(175.0mmol)的溴和300ml的二氯甲烷并向反应烧瓶缓慢滴注,在常温条件下进行整夜搅拌。反应结束后,向反应物投入碳酸氢钠水溶液并搅拌,利用二氯甲烷和蒸馏水提取。有机层在进行无水硫酸镁处理后过滤并进行减压浓缩。利用硅胶层析进行分离纯化来制备了21.5g(72%)的褐色固体中间体化合物[1-1]。
制备中间体化合物[1-2]
在氮气氛下,向1L的反应烧瓶投入20.5g(86.1mmol)的中间体化合物[1-1]、11g(90.4mmol)的苯基硼酸、1g(0.8mmol)的四(三苯基膦)钯以及205ml的1,4-二恶烷并提升温度。在60℃的温度条件下投入86.1ml的2M碳酸钾水溶液,进行整夜回流搅拌。在反应结束后,冷却至室温后利用乙酸乙酯、蒸馏水提取,有机层在进行无水硫酸镁处理后过滤。对滤液进行减压浓缩,利用乙酸乙酯和己烷以硅胶层析进行分离纯化。从而制备了16.3g(80%)的米色固体中间体化合物[1-2]。
制备中间体化合物[1-3]
在氮气氛下,向1L的反应烧瓶投入14.8g(62.9mmol)的中间体化合物[1-2]和123g(1064.4mmol)的吡啶盐酸盐,并进行整夜回流搅拌。在反应结束后,利用四氢呋喃、蒸馏水提取,有机层在进行无水硫酸镁处理后过滤并进行减压浓缩。利用丙酮、己烷重结晶化来制备了10.8g(78%)的米色固体中间体化合物[1-3]。
制备中间体化合物[1-4]
在氮气氛下,向1L的反应烧瓶投入10.8g(48.7mmol)的中间体化合物[1-3]、430ml的二氯甲烷、8ml(97.4mmol)的吡啶并搅拌。在0℃的温度条件下缓慢滴注12ml(73.0mmol)的无水三氟甲基砜,在常温条件下进行整夜搅拌。在反应结束后,通过投入400ml的蒸馏水来提取,之后,在有机层进行无水硫酸镁处理后进行二氧化硅(silica)过滤。通过对滤液进行减压浓缩来制备了13.8g(80%)的淡黄色固体中间体化合物[1-4]。
制备中间体化合物[1-5]
在氮气氛下,向500ml的反应烧瓶投入12.8g(36.2mmol)的中间体化合物[1-4]、11.9g(47.1mmol)的双(频哪醇合)二硼、10.7g(108.6mmol)的乙酸钾、0.3g(1.1mmol)的乙酸钯、1.0g(2.2mmol)的2-二环己基磷-2,4,6-三异丙基联苯(XPhos)、180ml的四氢呋喃并进行整夜回流搅拌。在反应结束后,利用二氯甲烷、蒸馏水提取,在有机层进行无水硫酸镁处理并进行减压浓缩。利用乙酸乙酯和己烷且通过硅胶层析进行分离纯化,从而制备了10.2g(85%)的白色固体中间体化合物[1-5]。
制备化合物[1]
在氮气氛下,向500ml的反应烧瓶投入10.2g(30.8mmol)的中间体化合物[1-5]、8.6g(32.3mmol)的2-氯-4,6-二苯基-1,3,5-三嗪、0.7g(0.6mmol)的四(三苯基膦)钯、140ml的1,4-二恶烷并升温。在60℃的温度条件下添加30.8ml的2M碳酸钾水溶液并进行4小时的回流搅拌。在反应结束后过滤,利用丙酮、甲醇、甲苯进行重结晶化,从而制备了6.8g(51%)的白色固体的目标化合物[1]。
合成例2:制备化合物[13]
反应式2:
制备化合物[13]
代替2-氯-4,6-二苯基-1,3,5-三嗪使用2-氯-4-(4-萘-2-基)苯基)-6-苯基-1,3,5-三嗪,除此之外,通过与上述化合物[1]的制备方法相同的方法进行合成来制备了12.1g(70%)的白色固体的目标化合物[13]。
合成例3:制备化合物[71]
反应式3:
制备中间体化合物[71-1]
在氮气氛下,向500ml的反应烧瓶投入10g(28.3mmol)的中间体化合物[1-4]、4.6g(29.7mmol)的4-氯苯基硼酸、0.7g(0.6mmol)的四(三苯基膦)钯、150ml的1,4-二恶烷并升温。在60℃的温度条件下添加28.3ml的2M碳酸钾水溶液,进行3小时的回流搅拌。利用乙酸乙酯、蒸馏水提取,在有机层进行无水硫酸镁处理并进行减压浓缩。利用二氯甲烷和己烷且通过硅胶层析进行分离纯化,从而制备了7.7g(86%)的白色固体中间体化合物[71-1]。
制备中间体化合物[71-2]
在氮气氛下,向500ml的反应烧瓶投入7.7g(24.3mmol)的中间体化合物[71-1]、8.0g(31.6mmol)的双(频哪醇合)二硼、7.1g(72.9mmol)的乙酸钾、0.2g(0.7mmol)的乙酸钯、0.7g(1.4mmol)的2-二环己基磷-2,4,6-三异丙基联苯、154ml的四氢呋喃并进行整夜回流搅拌。在反应结束后,利用二氯甲烷、蒸馏水提取,在有机层进行无水硫酸镁处理并进行减压浓缩。利用乙酸乙酯和己烷以硅胶层析进行分离纯化,从而制备了8.0g(81%)的白色固体中间体化合物[71-2]。
制备化合物[71]
在氮气氛下,向500ml的反应烧瓶投入8.0g(19.7mmol)的中间体化合物[71-2]、7.1g(20.7mmol)的2-(4-联苯)-4-氯-6-苯基-1,3,5-三嗪、0.5g(0.4mmol)的四(三苯基膦)钯、120ml的1,4-二恶烷并升温。在60℃的温度条件下添加19.7ml的2M碳酸钾水溶液并进行4小时的回流搅拌。在反应结束后过滤,利用丙酮、甲醇、甲苯进行重结晶化,从而制备了5.2g(45%)的白色固体的目标化合物[71]。
合成例4:制备化合物[80]
反应式4:
制备化合物[80]
代替2-(4-联苯)-4-氯-6-苯基-1,3,5-三嗪使用2-氯-4-(4-萘-1-基)苯基)-6-苯基-1,3,5-三嗪,除此之外,通过与上述化合物[71]的制备方法相同的方法进行合成来制备了7.2g(57%)的白色固体的目标化合物[80]。
合成例5:制备化合物[91]
反应式5:
制备化合物[91]
代替2-(4-联苯)-4-氯-6-苯基-1,3,5-三嗪使用3'-(4-氯-6-苯基-1,3,5-三嗪-2-基)-[1,1'-联苯]-4-腈,除此之外,通过与上述化合物[71]的制备方法相同的方法进行合成来制备了6.1g(50%)的白色固体的目标化合物[91]。
合成例6:制备化合物[149]
反应式6:
制备中间体化合物[149-1]
代替4-氯苯基硼酸使用3-氯苯基硼酸,除此之外,通过与上述中间体化合物[71-1]的制备方法相同的方法进行合成来制备了7.3g(82%)的白色固体中间体化合物[149-1]。
制备中间体化合物[149-2]
代替中间体化合物[71-1]使用中间体化合物[149-1],除此之外,通过与上述中间体化合物[71-2]的制备方法相同的方法进行合成来制备了8.1g(86%)的白色固体中间体化合物[149-2]。
制备化合物[149]
代替中间体化合物[71-2]使用中间体化合物[149-2],代替2-(4-联苯)-4-氯-6-苯基-1,3,5-三嗪使用2-氯-4-(4-萘-1-基)苯基)-6-苯基-1,3,5-三嗪,除此之外,通过与上述化合物[71]的制备方法相同的方法进行合成来制备了6.4g(50%)的白色固体目标化合物[149]。
合成例7:制备化合物[178]
反应式7:
制备化合物[178]
代替2-氯-4-(4-萘-1-基)苯基)-6-苯基-1,3,5-三嗪使用2-氯-4-(二苯并呋喃-3-基)-6-苯基-1,3,5-三嗪,除此之外,通过与上述化合物[149]的制备方法相同的方法进行合成来制备了7.1g(59%)的白色固体目标化合物[178]。
合成例8:制备化合物[210]
反应式8:
制备中间体化合物[210-1]
代替4-氯苯基硼酸使用2-氯苯基硼酸,除此之外,通过与上述中间体化合物[71-1]的制备方法相同的方法进行合成来制备了6.7g(75%)的白色固体中间体化合物[210-1]。
制备中间体化合物[210-2]
代替中间体化合物[71-1]使用中间体化合物[210-1],除此之外,通过与上述中间体化合物[71-2]的制备方法相同的方法进行合成来制备了7.2g(83%)的白色固体中间体化合物[210-2]。
制备化合物[210]
代替中间体化合物[71-2]使用中间体化合物[210-2],除此之外,通过与上述化合物[71]的制备方法相同的方法进行合成来制备了4.2g(41%)的白色固体的目标化合物[210]。
合成例9:制备化合物[220]
反应式9:
制备化合物[220]
代替2-(4-联苯)-4-氯-6-苯基-1,3,5-三嗪使用2-氯-4-(4-(萘-1-基)苯基)-6-苯基-1,3,5-三嗪,除此之外,通过与上述化合物[210]的制备方法相同的方法进行合成来制备了4.9g(44%)的白色固体的目标化合物[220]。
合成例10:制备化合物[280]
反应式10:
制备中间体化合物[280-1]
在氮气氛下,向500ml的反应烧瓶投入20g(60.4mmol)的中间体化合物[1-5]、18.8g(60.4mmol)的4,4'-二溴-1,1'-联苯、1.4g(1.2mmol)的四(三苯基膦)钯、200ml的1,4-二恶烷并升温。在60℃的温度条件下,添加60.4ml的2M碳酸钾水溶液并进行6小时的回流搅拌。在反应结束后过滤,利用二氯甲烷和己烷且通过硅胶层析进行分离纯化,从而制备了17.6g(67%)的白色固体中间体化合物[280-1]。
制备中间体化合物[280-2]
代替中间体化合物[71-1]使用了中间体化合物[280-1],除此之外,通过与上述中间体化合物[71-2]的制备方法相同的方法进行合成来制备了17.5g(90%)的白色固体中间体化合物[280-2]。
制备化合物[280]
代替中间体化合物[71-2]使用中间体化合物[280-2],代替2-(4-联苯)-4-氯-6-苯基-1,3,5-三嗪使用2-氯-4,6-二苯基-1,3,5-三嗪,除此之外,通过与上述化合物[71]的制备方法相同的方法进行合成来制备了12.6g(59%)的白色固体的目标化合物[280]。
合成例11:制备化合物[283]
反应式11:
制备中间体化合物[283-1]
代替4,4’-二溴-1,1’-联苯使用2,2’-二溴-1,1’-联苯,除此之外,通过与上述中间体化合物[280-1]的制备方法相同的方法进行合成来制备了18.8g(72%)的白色固体中间体化合物[283-1]。
制备中间体化合物[283-2]
代替中间体化合物[280-1]使用中间体化合物[283-1],除此之外,通过与上述中间体化合物[280-2]的制备方法相同的方法进行合成来制备了16.9g(87%)的白色固体中间体化合物[283-2]。
制备化合物[283]
代替中间体化合物[280-2]使用中间体化合物[283-2],除此之外,通过与上述化合物[280]的制备方法相同的方法进行合成来制备了10.7g(50%)的白色固体的目标化合物[283]。
合成例12:制备化合物[308]
反应式12:
制备中间体化合物[308-1]
代替6-甲氧基喹啉使用2-氯-6-甲氧基喹啉,除此之外,通过与上述中间体化合物[1-1]的制备方法相同的方法进行合成来制备了21.4g(76%)的黄色固体中间体化合物[308-1]。
制备中间体化合物[308-2]
在氮气氛下,向1L的反应烧瓶投入21.4g(78.5mmol)的中间体化合物[308-1]、9.6g(78.5mmol)的苯基硼酸、1.8g(1.6mmol)的四(三苯基膦)钯及214ml的1,4-二恶烷并升温。在60℃的温度条件下投入78.5ml的2M碳酸钾水溶液并进行整夜回流搅拌。反应结束后,冷却至室温后,利用乙酸乙酯、蒸馏水提取,在有机层进行无水硫酸镁处理后过滤。对滤液进行减压浓缩,利用乙酸乙酯和己烷且通过硅胶层析进行分离纯化。制备了14.4g(68%)的米色固体中间体化合物[308-2]。
制备中间体化合物[308-3]
在氮气氛下,向1L的反应烧瓶投入14.4g(53.4mmol)的中间体化合物[308-2]、9.6g(56.1mmol)的2-萘基硼酸、1.3g(1.1mmol)的四(三苯基膦)钯及144ml的1,4-二恶烷并升温。在60℃的温度条件下投入53.4ml的2M碳酸钾水溶液并进行整夜回流搅拌。反应结束后,冷却至室温后,利用乙酸乙酯、蒸馏水提取,在有机层进行无水硫酸镁处理后过滤。对滤液进行减压浓缩,利用乙酸乙酯和己烷且通过硅胶层析进行分离纯化。制备了15.8g(82%)的米色固体中间体化合物[308-3]。
制备中间体化合物[308-4]
代替中间体化合物[1-2]使用中间体化合物[308-3],除此之外,通过与上述中间体化合物[1-3]的制备方法相同的方法进行合成来制备了11.4g(75%)的米色固体中间体化合物[308-4]。
制备中间体化合物[308-5]
代替中间体化合物[1-3]使用中间体化合物[308-4],除此之外,通过与上述中间体化合物[1-4]的制备方法相同的方法进行合成来制备了13.5g(86%)的淡黄色固体中间体化合物[308-5]。
制备中间体化合物[308-6]
代替中间体化合物[1-4]使用中间体化合物[308-5],除此之外,通过与上述中间体化合物[1-5]的制备方法相同的方法进行合成来制备了10.4g(81%)的白色固体中间体化合物[308-6]。
制备化合物[308]
代替中间体化合物[1-5]使用中间体化合物[308-6],代替2-氯-4,6-二苯基-1,3,5-三嗪使用2-(4-溴苯基)-4,6-二苯基-1,3,5-三嗪,除此之外,通过与上述化合物[1]的制备方法相同的方法进行合成来制备了9.0g(62%)的白色固体的目标化合物[308]。
根据上述合成例1至12的制备方法制备了化合物1至化合物324,在下述表2示出对于所制备的各个化合物进行核磁共振氢谱(
表2
比较例化合物
比较例1
将由上述化学式f表示的化合物f用作荧光蓝色主剂,将由上述化学式g表示的化合物g用作荧光蓝色掺杂剂,将2-TNATA(4,4',4"-三(N-萘-2-基)-N-苯氨基)-三苯胺;4,4',4"-tris(N-naphthalen-2-yl)-N-phenylamino)-triphenylamine)用作空穴注入层物质,将α-NPD(N,N’-二萘-1-基)-N,N'-二苯基联苯胺;N,N'-di(naphthalene-1-yl)-N,N’-(diphenylbenzidine)用作空穴传输层物质,从而制备了具有如下结构的有机电致发光器件:氧化铟锡(ITO)/2-TNATA(60nm)/α-NPD(30nm)/化合物f+化合物g(30nm)/Alq
将康宁(Corning)公司的15Ω/cm
比较例2~5
在上述比较例1中,在电子传输层代替Alq
将此称为比较例2至5。
比较例6
在发光层的上部将5nm的上述BCP作为空穴屏蔽层化合物进行蒸镀来形成空穴阻挡层,在上述空穴阻挡层的上部真空蒸镀化合物b来形成电子传输层,除此之外,通过与上述比较例1相同的方法制备了有机电致发光器件。将其称为比较例6。
实施例1~9
代替用作电子传输层的Alq
实施例10~12
将化合物b用作电子传输层,代替用作空穴阻挡层的化合物BCP,对如上述表1所示的化合物210、220或283进行升华纯化过程并分别用作空穴屏蔽层,除此之外,通过与上述比较例6相同的方法制备了有机电致发光器件。将其分别称为实施例10至12。
实施例13
代替b,将化合物71用作电子传输层,代替用作空穴阻挡层的化合物BCP,对化合物210进行升华纯化过程并用作空穴屏蔽层,除此之外,通过与上述比较例6相同的方法制备了有机电致发光器件。将其称为实施例13。
评价例1:比较例1~6及实施例1~13的发光特性及寿命评价
对于比较例1~6及实施例1~13,利用Keithley sourcemeter“2400”、KONIKAMINOLTA“CS-2000”进行发光峰值、发光效率评价,
利用McScience公司的M6000S寿命测量装置以(L0)1000nit的初始亮度为基准分别测量亮度(L)达到97%的时间(LT97),在下述表3及图1至3示出其结果。
表3
如上述表3所示,相比于比较例1~5,实施例1~9示出低电压驱动及得到提高的发光特性。并且,当包括空穴屏蔽层时,也示出低电压驱动及得到提高的发光特性及寿命特性。
图1为示出对在实施例1~4及比较例1~5中制备的有机电致发光器件进行上述测量结果的寿命特性评价曲线图。
图2为示出对在实施例5~9中制备的有机电致发光器件进行上述测量结果的寿命特性评价曲线图。
图3为示出对在实施例10~13及比较例6中制备的有机电致发光器件进行上述测量结果的寿命特性评价曲线图。
如上述表3所示,相比于比较例1~6,实施例1至13示出得到提高的寿命特性。尤其,示出位于5、6号位置的取代的三嗪基喹啉衍生物的上述有机发光化合物示出优秀的性能和寿命。
以上,详细说明了本发明的优选实施例,但本发明的发明要求保护范围并不限定于此,属于普通技术人员利用发明要求保护范围所定义的本发明的基本概念的多个变形及改良形态也属于本发明的发明要求保护范围。
- 一种有机化合物,包含该化合物的有机电致发光器件材料及包含该材料的有机电致发光器件
- 一种有机化合物,包含该化合物的有机电致发光器件材料及包含该材料的有机电致发光器件