依达拉奉前体药物化合物及其在治疗或改善神经退行性或运动神经元疾病中的医药用途
文献发布时间:2023-06-19 10:44:55
技术领域
本发明涉及依达拉奉(edaravone)化合物的新型前体药物(prodrug)或其药学上可接受的盐,依达拉奉已知可用于治疗或改善神经退行性和运动神经元疾病,例如卢-格里格氏病(Lou Gehrig's disease)。本发明还涉及包含这种新型前体药物或其药学上可接受的盐作为有效成分的药物组合物。本发明还涉及使用这种前体药物或其药学上可接受的盐的医药用途。
背景技术
目前,用于退行性脑疾病或运动神经元疾病的治疗剂的临床应用非常有限,这些疾病包括阿尔茨海默氏病(Alzheimer's disease)、帕金森氏病(Parkinson's disease)、亨廷顿氏病(Huntington's disease)、卢-格里格氏病(Lou Gehrig's disease,Amyotrophic lateral sclerosis)或多发性硬化症(multiple sclerosis)。为了这种疾病的治疗或症状改善,对于每种疾病已经采取了各种方法,而可以普遍应用于该疾病组的治疗机制是抑制由病原性蛋白质引起的神经细胞氧化损伤的方法。也就是说,氧自由基(radical oxygen species,ROS)的过量产生会导致神经毒性,而抗氧化剂可通过减少神经氧化损伤来减少神经退化。
另一方面,依达拉奉是一种于2017年在美国被新批准用于治疗卢-格里格氏病的药物,其在临床试验中改善卢-格里格氏病的功能评定量表(revised ALS functionalrating scale,ALSFRS-R)并显著降低了脑脊液(cerebrospinal fluid,CSF)中3-硝基酪氨酸(3-nitrotyrosine)的水平(药物治疗专家意见(Expert Opinion onPharmacotherapy),2017,18(7),735)的作用已得到证实。尽管依达拉奉对卢-格里格氏的作用机理尚未得到准确的确定,但考虑到氧化应激相关的神经元细胞死亡的假说,推测依达拉奉的抗氧化功能是显示卢-格里格氏病的治疗作用的机制(衰老神经科学前沿(Frontiers in Aging Neuroscience),2017,9,68)。
另一方面,阿尔茨海默氏病显示出β-淀粉样蛋白(amyloid-β)的积累和由此引起的病理学特征。据报道,依达拉奉抑制β-淀粉样蛋白的沉积以及由此氧化损伤,并抑制疾病进展(美国国家科学院院刊(Proccedings of the National Academy of Sciences ofthe United States of America),2015,112(16),5225)。也就是说,在使用诱发阿尔茨海默氏病的APPswe/PS1小鼠的动物模型试验中腹腔内施用依达拉奉时,证实了β-淀粉样蛋白沉积减少和tau蛋白磷酸化减少、神经炎症和神经元丢失减少的同时,还证实了行为学改善效果。
此外,帕金森氏病显示出在作为运动神经中枢的基底核中缺乏多巴胺物质。然而,将依达拉奉用于作为慢性帕金森病诱发动物的鱼藤酮大鼠模型(chronic rotenone ratmodel)时,肌肉僵硬现象(catalepsy)、多巴胺神经元的退化等均得到了极大的改善。事实证明,这是由于依达拉奉大大降低了中脑中存在的氧自由基(PLoS One,2011,6(6),e20677)。
依达拉奉目前以静脉注射(IV infusion)的形式商品化,通常以28天(每次60mg)的重复周期为基础向患者给药。换句话说,在初期给药14天后,经过14天的休止期(restperiod),然后在14天中再次给药10天后,再经过14天的休止期。但是,从患者的角度来看,每次给药都要去医院并接受一个小时的注射是非常麻烦的过程。尤其,因卢-格里格氏病的特征,考虑到不便的患者的移动、注射给药、频繁的给药计划,即使该药物具有临床实用性,但在患者的便利性方面还是非常不足的。
这种不便可以通过例如将静脉注射剂改变成弹丸注射剂或进一步开发为口服制剂来解决。但是,由于依达拉奉的低溶解度(1.85mg/ml)、口服吸收率低(F
另一方面,中国专利公开号102190622提出了具有特定结构的前体药物(prodrugs),其是通过氨基甲酸酯官能团与依达拉奉连接的哌嗪衍生物。并且,将这些化合物经口服给药或注射到动物模型中以测量其抗氧化作用。但是,上述专利未公开化合物体内给药的药代动力学方面的内容,并且也未公开或暗示从前体药物产生的依达拉奉的血药浓度。
本发明人通过亲自实验确认的结果,中国专利中公开的前体药物不足以改善依达拉奉的生物利用度,因此,本发明人确定需要一种具有显著改善的吸收率和生物利用度的药物。
发明内容
发明要解决的问题
因此,本发明要解决的问题是提供具有改善的吸收率、生物利用度等的依达拉奉前体药物、包含这种前体药物的药物组合物以及这种前体药物在治疗或改善神经退行性和/或运动神经元疾病中的医药用途。
用于解决问题的手段
为了解决上述问题,本发明提供由以下化学式1表示的化合物或其药学上可接受的盐。
[化学式1]
在上述化学式1中,
R
R
本发明人通过制备和评价各种前体药物,证实了上述化合物或其药学上可接受的盐在吸收率和生物利用度方面表现出非常优异的效果,并且除了生物利用度方面,还证实了其表现出作为前提药物的优异的物理性质或特性,从而完成了本发明。
在本发明的一个实施方式中,优选地,所述化合物为3-甲基-1-苯基-1H-吡唑-5-基4-乙酰基哌嗪-1-羧酸酯(3-methyl-1-phenyl-1H-pyrazol-5-yl 4-acetylpiperazine-1-carboxylate);(S)-3-甲基-1-苯基-1H-吡唑-5-基4-乙酰基-2-甲基哌嗪-1-羧酸酯((S)-3-methyl-1-phenyl-1H-pyrazol-5-yl 4-acetyl-2-methylpiperazine-1-carboxylate);(R)-3-甲基-1-苯基-1H-吡唑-5-基4-乙酰基-2-甲基哌嗪-1-羧酸酯((R)-3-methyl-1-phenyl-1H-pyrazol-5-yl 4-acetyl-2-methylpiperazine-1-carboxylate);3-甲基-1-苯基-1H-吡唑-5-基4-(环己烷羰基)哌嗪-1-羧酸酯(3-Methyl-1-phenyl-1H-pyrazol-5-yl 4-(cyclohexanecarbonyl)piperazine-1-carboxylate);3-甲基-1-苯基-1H-吡唑-5-基4-苯甲酰哌嗪-1-羧酸酯(3-Methyl-1-phenyl-1H-pyrazol-5-yl 4-benzoylpiperazine-1-carboxylate);3-甲基-1-苯基-1H-吡唑-5-基4-(2-氨基乙酰基)哌嗪-1-羧酸酯(3-Methyl-1-phenyl-1H-pyrazol-5-yl 4-(2-aminoacetyl)piperazine-1-carboxylate);(S)-3-甲基-1-苯基-1H-吡唑-5-基4-(2-氨基丙酰基)哌嗪-1-羧酸酯((S)-3-methyl-1-phenyl-1H-pyrazol-5-yl 4-(2-aminopropanoyl)piperazine-1-carboxylate);(S)-3-甲基-1-苯基-1H-吡唑-5-基4-(2-氨基-3-羟基丙酰基)哌嗪-1-羧酸酯((S)-3-methyl-1-phenyl-1H-pyrazol-5-yl 4-(2-amino-3-hydroxypropanoyl)piperazine-1-carboxylate);(R)-3-甲基-1-苯基-1H-吡唑-5-基4-(2-氨基-3-巯基丙酰基)哌嗪-1-羧酸酯((R)-3-methyl-1-phenyl-1H-pyrazol-5-yl 4-(2-amino-3-mercaptopropanoyl)piperazine-1-carboxylate);(S)-3-甲基-1-苯基-1H-吡唑-5-基4-(2-氨基-3-甲基丁酰基)哌嗪-1-羧酸酯((S)-3-methyl-1-phenyl-1H-pyrazol-5-yl 4-(2-amino-3-methylbutanoyl)piperazine-1-carboxylate);(R)-3-甲基-1-苯基-1H-吡唑-5-基4-((S)-2-氨基-3-甲基丁酰基)-2-甲基哌嗪-1-羧酸酯((R)-3-methyl-1-phenyl-1H-pyrazol-5-yl 4-((S)-2-amino-3-methylbutanoyl)-2-methylpiperazine-1-carboxylate);(S)-3-甲基-1-苯基-1H-吡唑-5-基4-((S)-2-氨基-3-甲基丁酰基)-2-甲基哌嗪-1-羧酸酯((S)-3-methyl-1-phenyl-1H-pyrazol-5-yl 4-((S)-2-amino-3-methylbutanoyl)-2-methylpiperazine-1-carboxylate);(R)-3-甲基-1-苯基-1H-吡唑-5-基4-((S)-2-氨基-3-甲基丁酰基)-2-乙基哌嗪-1-羧酸酯((R)-3-methyl-1-phenyl-1H-pyrazol-5-yl 4-((S)-2-amino-3-methylbutanoyl)-2-ethylpiperazin-1-carboxylate);(S)-3-甲基-1-苯基-1H-吡唑-5-基4-((S)-2-氨基-3-甲基丁酰基)-3-甲基哌嗪-1-羧酸酯((S)-3-methyl-1-phenyl-1H-pyrazol-5-yl 4-((S)-2-amino-3-methylbutanoyl)-3-methylpiperazin-1-carboxylate);(S)-3-甲基-1-苯基-1H-吡唑-5-基4-(2-氨基-3,3-二甲基丁酰基)哌嗪-1-羧酸酯((S)-3-methyl-1-phenyl-1H-pyrazol-5-yl 4-(2-amino-3,3-dimethylbutanoyl)piperazine-1-carboxylate);(S)-3-甲基-1-苯基-1H-吡唑-5-基4-(2-氨基-4-(甲硫基)丁酰基)哌嗪-1-羧酸酯((S)-3-methyl-1-phenyl-1H-pyrazol-5-yl 4-(2-amino-4-(methylthio)butanoyl)piperazine-1-carboxylate);(S)-3-甲基-1-苯基-1H-吡唑-5-基4-(2-氨基-3-苯基丙酰基)哌嗪-1-羧酸酯((S)-3-methyl-1-phenyl-1H-pyrazol-5-yl 4-(2-amino-3-phenylpropanoyl)piperazine-1-carboxylate);(S)-3-甲基-1-苯基-1H-吡唑-5-基4-(2,4-二氨基-4-氧代丁酰基)哌嗪-1-羧酸酯((S)-3-methyl-1-phenyl-1H-pyrazol-5-yl 4-(2,4-diamino-4-oxobutanoyl)piperazine-1-carboxylate);或者(S)-3-甲基-1-苯基-1H-吡唑-5-基4-(2,6-二氨基己酰基)哌嗪-1-羧酸酯((S)-3-methyl-1-phenyl-1H-pyrazol-5-yl 4-(2,6-diaminohexanoyl)piperazine-1-carboxylate)。
在本说明书中,如果使用术语“Cx-Cy”时,则表示碳原子数为x至y。例如,(C
本发明中使用的术语“烷基”包括直链和支链型。
在本说明书中,术语“一种或多种药学上可接受的盐”是指由基于本发明的化合物的特定取代基的相对无毒的酸或碱来制备的活性化合物的盐。当化合物包含相对碱性的基团时,通过使中性化合物与足够量的所需酸和纯或合适的惰性(inert)溶剂接触可获得酸加成盐。药学上可接受的酸加成盐不仅包括衍生自相对无毒的有机酸的盐,包括但不限于乙酸、丙酸、异丁酸、草酸(oxalic)、马来酸(maleic)、丙二酸(malonic)、苯甲酸、琥珀酸、辛二酸(suberic)、富马酸(fumaric)、扁桃酸、邻苯二甲酸(phthalic)、苯磺酸(benzenesulfonic)、对甲苯磺酸(tolylsulfonic)、柠檬酸、酒石酸、甲磺酸(methanesulfonic)以及其类似物,还包括衍生自无毒的无机酸的烟,包括但不限于氯化氢、溴化氢、硝酸、碳酸、一氢碳酸(monohydrogencarbonic)、磷酸(phosphoric)、一氢磷酸、二氢磷酸、硫酸、一氢硫酸、碘化氢、亚磷酸(phosphorous)及其类似物。还包括氨基酸的盐,例如精氨酸(arginate)或其类似物,还包括有机酸的类似物,例如葡萄糖醛酸(glucuronic)或半乳糖醛酸(galacturonic)(例如,博格(Berge)等(1977)J.Pharm.Sci.66:1-19)。盐的其他实例在本领域众所周知的文献中公开,例如,公开于雷明顿药物科学,第18版,麦克出版社,伊斯顿PA(1990年)(Remington's PharmaceuticalSciences,l8th eds.,Mack Publishing,(Easton PA(1990))或者雷明顿药学的科学与实践,第19版,麦克出版社,伊斯顿PA(1995年)(Remington:The Science and Practice ofPharmacy,19th eds,Mack Publishing,Easton PA(1995))的内容。
在本说明书中,短语“本发明的化合物”不仅包括化学式1的各化合物,还包括其包合物(clathrates)、水合物、溶剂化物或(结晶)多晶型物。并且,即使术语“本发明的化合物”未提及其药学上可接受的盐,该术语也包括本发明的化合物的药学上可接受的盐。在一个实施方案中,本发明的化合物可以是立体异构体上的纯的化合物,例如,以基本不含立体异构体的(例如,85%ee以上、90%ee以上、95%ee以上、97%ee以上、99%ee以上)形式存在。即如果根据本发明的化学式1的化合物或其盐是互变(tautomeric)异构体和/或立体异构体(例如,几何异构体(geometrical isomer)和构象异构体(conformational isomers))时,则这些分离的异构体及其混合物也包括在本发明的范围内。如果本发明的化合物或其盐在其结构中具有不对称碳(asymmetric carbon)时,则它光学活性化合物及其外消旋混合物也包括在本发明的范围内。
在本说明书中,术语“多晶型物(polymorph)”是指本发明的化合物的固体晶体形式或其复合物(complex)。同一化合物的不同多晶型物可表现出不同的物理、化学和/或光谱性质。不同的物理性质包括但不限于稳定性(例如,对热或光的稳定性)、可压缩性和密度(在制剂化和产品制造中很重要)以及溶出度(会影响生物利用度)。稳定性的差异可能是由于化学反应性的变化(例如,不同的氧化作用,即使得剂型由一种多晶型物组成时变色快于由另一种多晶型物组成时变色)或机械特性(例如,以动力学上优选的的多晶型物形式保存的片剂碎片转化为热力学上更稳定的多晶型物)或两者(例如,一种多晶型物的片剂在高湿度下更容易分解)全部而导致的。多晶型物的不同物理性质会影响对其进行加工。例如,一种多晶型物由于如其颗粒的形状或尺寸分布可能比另一种多晶型物更容易形成溶剂化物,或者可能更难以过滤或除去杂质。
在本说明书中,术语“溶剂化物”是指包含通过非共价分子间力结合的化学计量或非化学计量的溶剂的根据本公开的化合物或其盐。优选的溶剂是挥发性的、无毒的溶剂,并且可以微量施用于人类。
在本说明书中,术语“水合物(hydrate)”是指包含通过非共价分子间力结合的化学计量或非化学计量的水的根据本公开的化合物或其盐。
在本说明书中,术语“包合物(clathrate)”是指包含能够捕获客体分子(例如,溶剂或水)的空间(例如,通道(channel))的晶格形式的化合物或其盐。
以本发明的上述化学式1表示的化合物可以例如通过以下途径合成。
所使用的第一种物质是被由烷基组成的R
本发明人通过改变依达拉奉化合物的化学结构,设计和制造了具有新结构的新型化合物,该新型结构能够在口服给药时显著提高口服吸收速率。体内口服新化合物后,依达拉奉物质的血药浓度最终与用于治疗或改善各种神经退行性和/或运动神经元疾病的药物功效成正比。这意味着通过基于静脉内施用后的依达拉奉的血液浓度,比较在口服施用根据本发明的新型化合物(即前体药物)之后经代谢过程而暴露于血液中的依达拉奉的浓度,来按比例地应用静脉内和口服剂量。
本发明人制备并评价了具有各种化学结构的化合物,以得到具有改善的口服吸收率的新型化合物。另外,在化合物的理化性能方面,考虑到药物的吸收基本上是通过被动扩散实现的,因此通过评价亲脂性来评价药物的吸收。为了防止由于过低的水溶性而引起的吸收减少,还评估了是否可以显示出最小的溶解度(minimum solubility)。
尤其,认为本发明的某些化合物可以用作肠膜中存在的跨膜转运蛋白(transmembrane transporter)中氨基酸转运蛋白(amino acid transporter)(如PepT1和LAT1等)的底物(substrate)。因此,尽管依达拉奉由于Pgp的释放而具有较低的生物利用度,但认为由于氨基酸转运蛋白的主动转运,使得本发明的某些化合物具有显著提高的生物利用度。然而,本发明不限于这种理论机制。
具体而言,例如,化学式1的化合物中,以下实施例10的化合物的药代动力学评价结果显示,与静脉给药依达拉奉相比,口服时的生物利用度为88.2%,由此确认了,还证实了与口服依达拉奉的生物利用度(4.9%)相比本发明化合物的口服生物利用度增加了约18倍。另外,在以下实施例15的化合物的药代动力学评价结果中,口服实施例15的化合物的生物利用度与静脉内施用的依达拉奉相比为60.7%,与口服依达拉奉的生物利用度(4.9%)相比显著提高了12倍。
同时,对于中国专利CN102190622的实施例2中公开的化合物,即将4-甲基-1-哌嗪甲酰依达拉奉酯(4-methyl-1-piperazinformyledarabone)(与本申请的参考例1化合物相同;3-甲基-1-苯基-1H-吡唑-5-基4-甲基哌嗪-1-羧酸酯),以与本发明中新提出的实施例化合物相同的方式实施和比较了药代动力学特性。其结果,该化合物的生物利用度为12.0%。因此,与CN102190622的已知物质相比,本发明提出的包括上述实施例10和15的化合物的化学式1的化合物,显示出大大改善的口服生物利用度。
在另一个实施方案中,本发明提供一种药物组合物,其包含根据本发明的化学式1的化合物或其药学上可接受的盐的治疗有效量,以及药学上可接受的载体。
在本说明书中,“有效量”是指减缓或最小化神经退行性和/或运动神经元疾病的本发明化合物的量;或足以在神经退行性和/或运动神经元疾病的治疗或管理中提供疗效的量。
作为药学上可接受的载体,例如,可以使用用于口服给药的载体或用于肠胃外给药的载体。口服载体可以包括乳糖、淀粉、纤维素衍生物、硬脂酸镁、硬脂酸等。另外,肠胃外给药的载体可以包括水、合适的油、生理盐水、葡萄糖水溶液和乙二醇水溶液等,并且还可以包括稳定剂和保存剂。合适的稳定剂可以是例如亚硫酸氢钠、亚硫酸钠或抗坏血酸的抗氧化剂。合适的保存剂可以是苯扎氯铵、对羟基苯甲酸甲酯或对羟基苯甲酸丙酯和氯丁醇。其他药学上可接受的载体可以参考下述文件中描述的载体,即雷明顿药物科学,第19版,麦克出版社,伊斯顿PA,1995年(Remington's Pharmaceutical Sciences,19th ed.,MackPublishing Company,Easton PA,1995)所记载的载体。
本发明的药物组合物可以通过任何给药途径施用于包括人在内的哺乳动物。它可以口服或肠胃外给药。但是,从本发明的化合物显示出优异的口服吸收性的方面来看,口服给药途径是更优选的。
作为肠胃外给药方法,例如包括静脉内、肌内、动脉内、髓内、鞘膜内、心内、经皮、皮下、腹腔内、鼻内、肠、局部、舌下或直肠给药,但不限于此。例如,本发明的药物组合物可以以可注射制剂的形式制备,并通过用30G的细针轻刺皮肤或直接将其涂敷到皮肤上的方法来施用。
本发明的药物组合物可以根据如上所述的给药途径配制成用于口服或肠胃外给药的制剂。
就口服制剂的而言,本发明的组合物可以使用本领域已知的方法来配制,例如可以是粉末、颗粒、片剂、丸剂、糖衣片剂、胶囊、液剂、凝胶、糖浆、浆液(slurry)、悬浮剂等等。例如,作为口服制剂,通过将活性成分与固体赋形剂混合,粉碎,添加合适的辅助剂并将其加工成颗粒混合物,从而可以制成片剂。合适的赋形剂的实例包括:糖类,包括乳糖、葡萄糖、蔗糖、山梨糖醇、甘露醇、木糖醇、赤藓糖醇和麦芽糖醇等;淀粉类,包括玉米淀粉、小麦淀粉、大米淀粉和马铃薯淀粉等;纤维素类,包括纤维素、甲基纤维素、羧甲基纤维素钠和羟丙基甲基纤维素等;明胶、聚乙烯吡咯烷酮等的填充剂。另外,在某些情况下,可以添加交联聚乙烯吡咯烷酮、琼脂、海藻酸或海藻酸钠作为崩解剂。此外,本发明的药物组合物可以进一步包含抗凝剂、润滑剂、湿润剂、香料、乳化剂和防腐剂。
就肠胃外给药的制剂而言,可以通过本领域已知的方法将其制成注射剂、霜剂、乳剂、外用药膏剂、油剂、保湿剂、凝胶剂、气雾剂和鼻吸入剂的形式。这些制剂均被记载在所有药物化学领域众所周知的材料“《雷明顿药物科学》,第15版,1975年,麦克出版社,伊斯顿,宾夕法尼亚州18042,第87章:布卢格,西摩(Remington's Pharmaceutical Science,15th Edition,1975.Mack Publishing Company,Easton,Pennsylvania 18042,Chapter87:Blaug,Seymour)”中。
本发明的药物组合物的总剂量可以是以单剂量(single dose)向患者给药,并且也可以通过分次治疗方案给药(fractionated treatment protocol),所述分次治疗方案以多次剂量(multiple dose)长期给药。本发明的药物组合物可以根据疾病的症状来改变活性成分的含量。本发明的组合物的优选总剂量可以是每kg患者体重每天约0.01μg至1000mg,最优选0.1μg至100mg。然而,本发明药物组合物的合适有效剂量,除了给药途径和给药次数之外,本领域技术人员还可考虑患者的年龄、体重、健康状况、性别、疾病的严重程度、饮食和排泄率等多种因素而确定。本发明的药物组合物只要具有本发明的效果,就不会特别限定于任何特定的制剂、给药途径和给药方法。
另外,本发明的药物组合物可以作为单独的治疗剂给药或与另一种治疗剂组合给药。当与其他治疗剂组合给药时,本发明的组合物和其他治疗剂可以同时、单独或依次进行给药。此时,另一种治疗剂可以是已知具有治疗或改善神经退行性和/或运动神经元疾病的作用的物质。当本发明的药物组合物与另一种治疗剂组合给药时,本发明的组合物和另一种治疗剂可以分别配制在单独的容器中,或者可以组合在同一制剂中。
为了将本发明提出的化合物施用于人体,通过使用如下表1所示的片剂来详细描述代表性的药学方法。以下给出的化合物A和化合物B是指在本发明中提出的作为用于治疗、改善或预防神经退行性和运动神经元疾病的活性成分的物质。
[表1]
本发明还提供了用于治疗或改善神经退行性或运动神经元疾病的药物组合物,其包含根据本发明的化学式1的化合物或其药学上可接受的盐作为活性成分。即本发明提供了根据本发明的化学式1的化合物或其药学上可接受的盐在治疗或改善神经退行性或运动神经元疾病中的医药用途。
在另一个实施方案中,本发明提供了一种用于治疗或改善神经退行性或运动神经元疾病的方法,该方法包括将治疗有效量的化学式1的化合物或其药学上可接受的盐施用于需要其的受试者的步骤。上述神经退行性或运动神经元疾病是阿尔茨海默氏病、帕金森氏病、亨廷顿氏病、卢-格里格氏病、多发性硬化症、肌张力障碍、脊髓性肌萎缩症、炎性神经病或酒精性痴呆。另一方面,上述受试者是人。在一实施方式中,所述治疗是预防性治疗(preventative treatment)。在另一个实施方案中,所述治疗是姑息治疗(palliativetreatment)。在另一个实施方案中,治疗是恢复性治疗(restorative treatment)。
发明效果
本发明提供对神经退行性或运动神经元疾病有效的治疗或改善的化合物,以其为有效成分的药物组合物,其医药用途以及包括对需要治疗或预防的对象给药的治疗方法。根据本发明的化合物或其药学上可接受的盐作为医药产品的活性成分,在如溶解度等各个方面均具有多种优点,尤其是在口服给药后具有优异的生物利用度。
附图说明
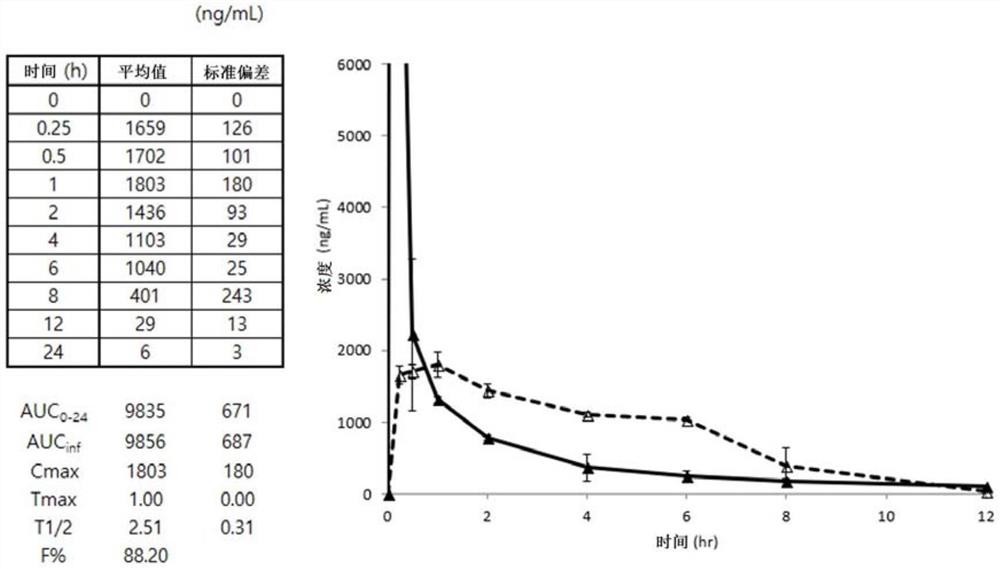
图1是作为比较物质的依达拉奉静脉给药和本发明的实施方式实施例10的化合物的单次口服给药后出现的依达拉奉的血液浓度随时间变化的图。在图1中,▲是依达拉奉静脉给药组的结果,△是实施例10化合物口服组的结果。
图2是作为比较物质的依达拉奉静脉给药和本发明的实施方式实施例15的化合物的单次口服给药后出现的依达拉奉的血液浓度随时间变化的图。在图2中,▲是依达拉奉静脉给药组的结果,△是实施例15化合物口服组的结果。
具体实施方式
将基于以下实施例更详细地描述本发明,但这并不意图限制本发明的范围。另外,本领域普通技术人员将能够在不脱离本发明的精神的范围内对本发明进行各种修改和变型。
首先,下面描述根据本发明的化学式1的化合物的实例。代表性的例子以及具体的制备步骤如下所述,具有不同取代基的化合物可通过相似的步骤制备。参考以下代表性实例,本领域普通技术人员将能够容易地制备具有不同取代基的化学式1的化合物。
参考例1:3-甲基-1-苯基-1H-吡唑-5-基4-甲基哌嗪-1-羧酸酯盐酸盐
将1.0g的1-甲基哌嗪溶解在10ml的二氯甲烷中,并添加1.2ml的吡啶(1.5eq.)。在氩气环境下将反应溶液冷却至0℃以下,然后向其中缓慢加入在15ml二氯甲烷中稀释的3.5g(1.2eq.)三光气。在室温搅拌2小时后,将其用25ml饱和盐水洗涤,并分离有机层。用无水硫酸镁干燥后,减压浓缩,得到黄色油状物。通过加入10ml乙腈完全溶解后,向其中加入1.74g(1.0eq.)的依达拉奉和9.76g(3eq.)的碳酸铯。在室温搅拌4小时后,使用硅藻土过滤反应溶液,并回收滤液并在减压下浓缩。将残余物用25ml乙酸乙酯溶解,用25ml饱和盐水洗涤,分离有机层,用无水硫酸镁干燥,并在减压下浓缩。将1.0ml浓盐酸水溶液和10ml乙酸乙酯加入到浓缩的残余物中,再次减压浓缩。向其中再次加入10ml乙酸乙酯,并在减压浓缩后,重复该过程3次以结晶,得到0.61g标题化合物(收率18.1%)。
制备例1:
在将苄基哌嗪-1-羧酸酯衍生物(benzyl piperazine-1-carboxylatederivative)溶于10倍体积的二氯甲烷中后,加入1.2当量的三乙胺和1.1当量的活性酯化合物。将混合物在氩气环境下于室温搅拌2小时,用饱和盐水洗涤,用无水硫酸镁干燥,并在减压下浓缩。将残余物溶解在四氢呋喃中,向其中添加5%的吸附在10wt%纯度的碳上的钯,然后在室温、常压氢气下搅拌。反应完成后,将反应溶液过滤后,将滤液回收进行减压浓缩。用硅胶柱色谱法(洗脱液:二氯甲烷和甲醇的混合溶液)纯化,得到酰化的哌嗪中间体。向其中添加10倍体积的二氯甲烷溶解,并添加1.5当量的吡啶。将反应溶液在氩气环境下冷却至0℃以下,然后缓慢滴加1.2当量的在15倍体积的二氯甲烷中稀释的三光气并搅拌。在保持0-5℃的条件下搅拌2小时后,用饱和盐水洗涤,并分离有机层。用无水硫酸镁干燥后,减压浓缩,得到黄色油状物。加入10倍体积的乙腈并完全溶解后,向其中加入1.0当量的依达拉奉和3.0当量的碳酸铯。在室温下搅拌,确认反应完成后,使用硅藻土过滤反应溶液,并回收滤液并在减压下浓缩。将残余物用10倍体积的乙酸乙酯溶解,用饱和盐水洗涤,分离有机层,用无水硫酸镁干燥,然后减压浓缩。将浓缩的残余物通过使用硅胶的柱色谱法纯化(洗脱液:乙酸乙酯和正己烷的混合溶液),得到目标化合物。
将苄基哌嗪-1-羧酸酯衍生物(benzyl piperazine-1-carboxylate derivative)溶解在5倍体积的二氯甲烷和5倍体积的N-甲基-2-吡咯烷酮(N-methyl-2-pyrrolidone,NMP)中,然后加入氨基被叔丁氧羰基(t-butoxycarbonyl,Boc)保护的1.0当量的氨基酸、1.1当量的二异丙基碳二亚胺(diisopropylcarbodiimide,DIPC)和1.2当量的三乙胺。将混合物在氩气环境下于室温搅拌2小时,用饱和盐水洗涤,用无水硫酸镁干燥,并在减压下浓缩。将残余物溶解在四氢呋喃中,5%的吸附在10wt%纯度的碳上的钯,然后在室温、常压氢气下搅拌。反应完成后,将反应溶液过滤,并将滤液回收进行减压浓缩。用硅胶柱色谱法(洗脱液:二氯甲烷和甲醇的混合溶液)纯化,得到酰化的哌嗪中间体。向其中添加10倍体积的二氯甲烷溶解,并添加1.5当量的吡啶。在氩气环境下,将反应溶液冷却至0℃以下,然后缓慢滴加1.2当量的稀释于15倍体积二氯甲烷中的三光气并搅拌。在室温搅拌2小时后,将其用饱和盐水洗涤并分离有机层。用无水硫酸镁干燥后,减压浓缩,得到黄色油状物。向其中加入10倍体积的乙腈并完全溶解后,向其中加入1.0当量的依达拉奉和3.0当量的碳酸铯。在室温下搅拌,确认反应完成后,使用硅藻土过滤反应溶液,并回收滤液并在减压下浓缩。将残余物用10倍体积的乙酸乙酯溶解,用饱和盐水洗涤,分离有机层,用无水硫酸镁干燥,然后减压浓缩。将浓缩的残余物通过使用硅胶的柱色谱法纯化(洗脱液:乙酸乙酯和正己烷的混合溶液)。向得到的所述中间体中加入5当量的溶解有4N盐酸的1,4-二恶烷(dioxane)溶液,使其完全溶解,在室温下搅拌30分钟,然后减压浓缩。向其中加入10倍体积的乙酸乙酯后,将其减压浓缩(重复3次)。最后,加入10倍体积的乙酸乙酯,并将获得的悬浮液过滤,以得到固体的目标化合物。
实施例1:3-甲基-1-苯基-1H-吡唑-5-基4-乙酰基哌嗪-1-羧酸酯
使用1.0g的苄基哌嗪-1-羧酸酯和0.36ml的乙酰氯,根据制备例1的方法获得了0.64g(42.9%)的标题化合物,为淡黄色固体。核磁共振分析和质量分析结果如下表2。
实施例2:(S)-3-甲基-1-苯基-1H-吡唑-5-基4-乙酰基-2-甲基哌嗪-1-羧酸酯
使用1.0g的(S)-苄基2-甲基哌嗪-1-羧酸酯和0.34ml的乙酰氯,根据制备例1的方法获得了0.60g(41.1%)的标题化合物,为淡黄色固体。核磁共振分析和质量分析结果如下表2。
实施例3:(R)-3-甲基-1-苯基-1H-吡唑-5-基4-乙酰基-2-甲基哌嗪-1-羧酸酯
使用1.0g的(R)苄基2-甲基哌嗪-1-羧酸酯和0.34ml的乙酰氯,根据制备例1的方法获得了0.72g(49.3%)的标题化合物,为淡黄色固体。核磁共振分析和质量分析结果如下表2。
实施例4:3-甲基-1-苯基-1H-吡唑-5-基4-(环己烷羰基)哌嗪-1-羧酸酯
使用1.0g的苄基哌嗪-1-羧酸酯和0.67ml的环己烷羰基氯,根据制备例1的方法获得了0.41g(22.8%)的标题化合物,为淡黄色固体。核磁共振分析和质量分析结果如下表2。
实施例5:3-甲基-1-苯基-1H-吡唑-5-基4-苯甲酰哌嗪-1-羧酸酯
使用1.0g的苄基哌嗪-1-羧酸酯和0.58ml的苯甲酰氯,根据制备例1的方法获得了0.84g(47.4%)的标题化合物,为米白色固体。核磁共振分析和质量分析结果如下表2。
实施例6:3-甲基-1-苯基-1H-吡唑-5-基4-(2-氨基乙酰基)哌嗪-1-羧酸酯盐酸盐
使用1.0g的苄基哌嗪-1-羧酸酯和0.80g的N-Boc-甘氨酸,根据制备例2的方法获得了0.42g(24.4%)的标题化合物,为米白色固体。核磁共振分析和质量分析结果如下表2。
实施例7:(S)-3-甲基-1-苯基-1H-吡唑-5-基4-(2-氨基丙酰基)哌嗪-1-羧酸酯盐酸盐
使用1.0g的苄基哌嗪-1-羧酸酯和0.86g的N-Boc-丙氨酸,根据制备例2的方法获得了0.38g(21.3%)的标题化合物,为米白色固体。核磁共振分析和质量分析结果如下表2。
实施例8:(S)-3-甲基-1-苯基-1H-吡唑-5-基4-(2-氨基-3-羟基丙酰基)哌嗪-1-羧酸酯盐酸盐
使用1.0g的苄基哌嗪-1-羧酸酯和1.45g的N-Boc-O-TBS-丝氨酸,根据制备例2的方法获得了0.29g(15.6%)的标题化合物,为米白色固体。核磁共振分析和质量分析结果如下表2。
实施例9:(R)-3-甲基-1-苯基-1H-吡唑-5-基4-(2-氨基-3-巯基丙酰基)哌嗪-1-羧酸酯盐酸盐
使用1.0g的苄基哌嗪-1-羧酸酯和1.71g的N-Boc-S-三异丙基甲硅烷基-半胱氨酸,根据制备例2的方法获得了0.18g(9.3%)的标题化合物,为淡黄色固体。核磁共振分析和质量分析结果如下表2。
实施例10:(S)-3-甲基-1-苯基-1H-吡唑-5-基4-(2-氨基-3-甲基丁酰基)哌嗪-1-羧酸酯盐酸盐
使用1.0g的苄基哌嗪-1-羧酸酯和0.99g的N-Boc-缬氨酸,根据制备例2的方法获得了0.39g(20.4%)的标题化合物,为米白色固体。核磁共振分析和质量分析结果如下表2。
实施例11:(R)-3-甲基-1-苯基-1H-吡唑-5-基4-((S)-2-氨基-3-甲基丁酰基)-2-甲基哌嗪-1-羧酸酯盐酸盐
使用1.0g的(R)苄基2-甲基哌嗪-1-羧酸酯和0.93g的N-Boc-缬氨酸,根据制备例2的方法获得了0.35g(18.8%)的标题化合物,为米白色固体。核磁共振分析和质量分析结果如下表2。
实施例12:(S)-3-甲基-1-苯基-1H-吡唑-5-基4-((S)-2-氨基-3-甲基丁酰基)-2-甲基哌嗪-1-羧酸酯盐酸盐
使用1.0g的(S)苄基2-甲基哌嗪-1-羧酸酯和0.93g的N-Boc-缬氨酸,根据制备例2的方法获得了0.30g(16.1%)的标题化合物,为米白色固体。核磁共振分析和质量分析结果如下表2。
实施例13:(R)-3-甲基-1-苯基-1H-吡唑-5-基4-((S)-2-氨基-3-甲基丁酰基)-2-乙基哌嗪-1-羧酸酯盐酸盐
使用1.0g的(R)苄基2-乙基哌嗪-1-羧酸酯和0.87g的N-Boc-缬氨酸,根据制备例2的方法获得了0.15g(8.3%)的标题化合物,为米白色固体。核磁共振分析和质量分析结果如下表2。
实施例14:(S)-3-甲基-1-苯基-1H-吡唑-5-基4-((S)-2-氨基-3-甲基丁酰基)-3-甲基哌嗪-1-羧酸酯盐酸盐
使用1.0g的(S)苄基3-甲基哌嗪-1-羧酸酯和0.93g的N-Boc-缬氨酸,根据制备例2的方法获得了0.36g(19.3%)的标题化合物,为米白色固体。核磁共振分析和质量分析结果如下表2。
实施例15:(S)-3-甲基-1-苯基-1H-吡唑-5-基4-(2-氨基-3,3-二甲基丁酰基)哌嗪-1-羧酸酯盐酸盐
使用1.0g的苄基哌嗪-1-羧酸酯和1.05g的N-Boc-t-亮氨酸,根据制备例2的方法获得了0.38g(19.2%)的标题化合物,为米白色固体。核磁共振分析和质量分析结果如下表2。
实施例16:(S)-3-甲基-1-苯基-1H-吡唑-5-基4-(2-氨基-4-(甲硫基)丁酰基)哌嗪-1-羧酸酯盐酸盐
使用1.0g的苄基哌嗪-1-羧酸酯和1.13g的N-Boc-蛋氨酸,根据制备例2的方法获得了0.28g(13.6%)的标题化合物,为米白色固体。核磁共振分析和质量分析结果如下表2。
实施例17:(S)-3-甲基-1-苯基-1H-吡唑-5-基4-(2-氨基-3-苯基丙酰基)哌嗪-1-羧酸酯盐酸盐
使用1.0g的苄基哌嗪-1-羧酸酯和1.20g的N-Boc-苯丙氨酸,根据制备例2的方法获得了0.35g(16.4%)的标题化合物,为米白色固体。核磁共振分析和质量分析结果如下表2。
实施例18:(S)-3-甲基-1-苯基-1H-吡唑-5-基4-(2,4-二氨基-4-氧代丁酰基)哌嗪-1-羧酸酯盐酸盐
使用1.0g的苄基哌嗪-1-羧酸酯和1.05g的N-Boc-天冬氨酸,根据制备例2的方法获得了0.15g(7.6%)的标题化合物,为棕色固体。核磁共振分析和质量分析结果如下表2。
实施例19:(S)-3-甲基-1-苯基-1H-吡唑-5-基4-(2,6-二氨基己酰基)哌嗪-1-羧酸二盐酸盐
使用1.0g的苄基哌嗪-1-羧酸酯和1.57g的N,N'-二-Boc-赖氨酸,根据制备例2的方法获得了0.20g(9.0%)的标题化合物,为棕色固体。核磁共振分析和质量分析结果如下表2。
上述实施例的核磁共振分析和质量分析结果在下表2中示出。
[表2]
实验例1:药代动力学评价
实施例和参考例化合物的药代动力学试验如下进行。即在向SD(Sprague-Dawley)大鼠(rat)单次口服测试化合物后,通过追踪通过代谢过程释放到血液中的依达拉奉的动力学(kinetics)并与标准物质比较,来验证本发明化合物的功效。具体而言,作为标准物质的依达拉奉分别通过静脉内和口服给药,而试验物质通过口服给药。然后,评价血液中依达拉奉的浓度。分别以相同的方式制备标准物质和试验物质,然后以0.1mmol/kg的剂量施用于大鼠,并在预定时间收集血液并分离血浆。用HPLC(XBridge C18色谱柱,沃特世(Waters),流动相0.1%甲酸:乙腈(30:70,%/%))和MS/MS(ESI阳性,MRM)进行药物分析。将每种市售标准溶液与大鼠血浆以9:1的比例混合,由此以5、50、100、500、100和5000ng/ml的浓度进行制备及校准。此外,QC样品是通过将大鼠血浆与QC用标准溶液以9:1的比例混合而成,浓度分别为100ng/ml、750ng/ml和2500ng/ml。在预处理方法中,将100μl血浆样品转移到离心管中,加入10μl内标准溶液和300μl甲醇后,混合约30秒。将试管在3000×g(4℃)下离心约5分钟,取上清液并转移至LC小瓶中,然后注入仪器中。另外,采用事先验证过的分析方法对大鼠血浆中有效成分即依达拉奉的浓度进行了定量。对于药代动力学参数,使用温诺林5.2(WinNonlin 5.2)(美国,Pharsight)程序,并通过非房室模型(Noncompartmentmodeling)(最佳拟合(best fit))计算AUC
实验后,标准物质和试验化合物口服后的每种生物利用度总结在下表3中。
[表3]
就代表性的实施例10化合物而言,平均AUC
如表3的结果所示,特别是实施例10、实施例19、实施例14和实施例15的化合物的口服生物利用度极其优异。
- 依达拉奉前体药物化合物及其在治疗或改善神经退行性或运动神经元疾病中的医药用途
- 用于神经退行性疾病和阿尔茨海默病的治疗中的用途的双重抑制剂化合物