用于在系谱内进行系谱富集和基于家族的分析的方法和系统
文献发布时间:2023-06-19 11:35:49
相关申请的交叉引用
本申请案要求2018年9月7日提交的美国临时专利申请号62/728,536的权益,该美国临时专利申请的内容据此以引用方式整体并入本文。
技术领域
本公开总体上涉及用于在大群体同期群中进行系谱富集的方法和系统。更具体地,本公开涉及用于使用测序数据在一级家族网络中识别患病者以富集系谱并进一步识别在系谱之内和系谱之间共隔离的变异性状对以将罕见的遗传变异与疾病和疾病易感性联系起来的系统和方法。
背景技术
临床研究人员一直在寻求识别导致疾病的致病变异。细胞基因组阵列和连锁小组的基因分型仍然是分别用于识别拷贝数变异和用于识别大型孟德尔(尤其是显性)疾病家族内的共隔离单倍型的有用方法。然而,仍不清楚在复杂疾病中探索致病变异的最佳方法。
通过系谱学追踪变异的传播是现代遗传学的基础。大多数遗传疾患为异质的,其范围为在致病中起作用的几个基因至多个基因。许多罕见疾患中的遗传缺陷仍然难以捉摸。使用经典定位克隆技术,需要大量患病的家族来识别致病基因应位于的区域,并且对于罕见疾患,这些家族并不总是可用的。此外,仅仅识别感兴趣的区域是不够的;该区域内的所有基因都必须进行测序,这可为非常费力的。随着下一代测序的出现,无需选择候选基因区域就可以研究患者的全基因组或外显子组。尽管我们现在可以在大型研究同期群中探索罕见的遗传变异并对其进行基因分型,但这些变异中的大多数将仅在少数个体中出现—在基于群体的遗传研究中,在单个个体中观察到了大于50%的变异—从而使得难以建立关联的证据。
在大型遗传同期群的全基因组扫描中研究罕见变异对这些异质性疾患的影响进一步尤其具有挑战性。序列变异的疾病因果关系的明确分配往往是不可能的,尤其是对于许多罕见、严重疾病病例的极低频变异。然而,如果识别出了一组共享给定遗传疾患的相关个体,则这种异质性将大大降低,从而允许专注于驱动系谱内患病个体中的特定表型隔离的单个基因和变异。
全基因组关联研究(GWAS)使得能够在整个人类基因组中无偏寻找疾病基因座的潜力,提供了遗传学中前所未有的研究机会。同时在许多受试者中查询数十万个单核苷酸多态性(SNP),在这些研究的设计和分析中提出了许多统计挑战。这种规模的基因分型需要新的方法来处理数据质量问题;同样,关联测试是针对成千上万个标记物计算的,其结果必须针对多个比较进行调整。这些问题的严重性提出了以下问题:对此类密集的SNP组进行基因分型的新技术能力是否将转化为对新型遗传病基因座的识别,或者技术进步是否仍未得到充分利用。至少有两种进行此类基因组范围关联研究的方法—基于群体的设计和基于家族的设计。
基于群体的研究具有数千名受试者的样本量(Szklo M.Epidemiologic Reviews(1998)20(1):81-90)。然而,这些研究昂贵、费时,并且由于样本量大而可能会遇到表型和基因型异质性(Sorlie和Wei.Journal of American College of Cardiology(2011)58(19):2010-3;Laird和Lange.Statistical Science(2009)24(4):388-397)。
当查询与感兴趣的表型共隔离的潜在中到大型效应的罕见变体时,基于家族的分析可为信息丰富的,并且使用基于群体的分析可能不容易检测到这些变异。基于家族的关联研究的一个主要益处是控制了由于群体分层而造成的混杂偏倚,尽管可能会丧失操纵力(Witte等人,American Journal of epidemiology(1999)149(8):693-705;Thomas等人,Cancer(2003)97(8):1894-1903)。
有许多大规模的测序计划用于确定数十万个身份不明的个体并对所述个体进行测序,例如DiscovEHR,UK Biobank,the US government’s All of US(PrecisionMedicine Initiative的一部分),TOPMed,ExAC/gnomAD,以及许多其他方面(Dewey等人,Science(2016);254,aaf6814;Sudlow等人,PLoSMed.(2015)12,e1001779;Collins等人,(2016)New England Journal of Medicine(2015)372,793-795;Lek等人,Nature(2016)536,285-291)。可以从如此大的蛋白质测序信息数据集构建系谱,研究者可以使用所述系谱来确定性状和疾患的遗传力和遗传模型。了解确切的系谱结构允许正确识别疾病遗传的遗传模式和利用需要或受益于真正系谱结构的强大遗传分析工具。然而,存在从去识别化的健康记录中直接获得准确的系谱记录挑战,排除了许多强大的基于家族的分析法。
可以使用紧密的成对关系来使用诸如PRIMUS和CLAPPER的工具直接从遗传数据重建系谱结构(Staples等人,American Journal of Human Genetics(2014)95,553-564以及Ko和Nielson.PLoS Genet.(2017)13,e1006963)。尽管估计的关系和系谱非常有用,但是存在关于在对估计的关系和系谱结构的不准确性敏感的分析中使用估计的关系和系谱存在很大的统计不确定性的问题。
虽然精确医学同期群可能不容易具有系谱信息,但可以直接从遗传数据获得信息丰富的系谱,以创建大型同期群而供进行传统孟德尔分析。识别针对具有感兴趣的表型的患病者富集的系谱可用于试图识别驱动这些表型的因果(罕见)变异,因为遗传原因更可能在家族单元内共享。定义系谱富集分析中使用的患病个体的集合可为关键的。因此,需要此类方法或系统来允许系谱富集。可以利用这些富集的系谱来帮助定义具有感兴趣表型的相关参与者的子集,然后检查这些子集以识别性状和疾病的遗传驱动因素。仍然需要用于系谱富集的改进的生物信息学工具,以识别潜在的信息丰富的系谱-表型配对,该潜在的信息丰富的系谱-表型配对使得能够实现大规模的传统孟德尔分析。
对产生富集系谱的方法和系统的探索可以指导药物探索科学家了解某些蛋白质及其变体在正常生理或疾病病因中所起的关键作用,并从生化和生物学角度阐明所述蛋白质及其变体的功能(Lele R.J.Assoc.Physicians India(2003)51:373-380)。
本文所述的方法和系统将提供富集系谱,该富集系谱可以导致识别出此类致病变异,并因此促进药物探索努力和临床研究努力。
发明内容
在一个示例性方面中,本公开提供了用于通过以下方式生成个体的一级网络的方法:基于同期群的测序数据生成个体的一级网络,将所述同期群中的个体识别为患病者或未患病者,以及创建包含所述患病者和所述未患病者的富集系谱。
在一些示例性实施方案中,所述用于生成富集系谱的方法可包括将系谱中的个体识别为患病者或未患病者,其中将具有至少一种二元性状的个体识别为患病者,并且将不具有所述至少一种二元性状的个体识别为未患病者,然后评估所述未患病和未患病个体的模式是否与孟德尔遗传模式(例如,常染色体显性遗传、常染色体隐性遗传、x连锁显性、x连锁隐性、或y连锁)一致。在一些特别的示例性实施方案中,可以使用国际疾病和相关健康问题统计分类(ICD)定义所述二元性状,ICD是由世界卫生组织(WHO)提供的医学分类列表,其包含疾病的代码、体征和症状、异常发现、主诉、社会环境以及伤害或疾病的外部原因。ICD的第九版本或第十版本可用于定义二元性状。在一个示例性实施方案中,没有电子健康记录数据的个体可用于该特定二元性状,或者具有关于该特定二元性状的冲突或不可靠数据的个体,无论病历中是否存在该特定二元性状,都可以确定是未知的患病者。
在一些示例性实施方案中,所述用于生成富集系谱的方法可包括将系谱中的个体识别为患病者或未患病者,其中将具有至少一种极端数量性状的个体识别为患病者,并且将不具有所述至少一种极端数量性状的个体识别为未患病者,然后评估所述未患病和未患病个体的模式是否与孟德尔遗传模式(例如,常染色体显性遗传、常染色体隐性遗传、x连锁显性、x连锁隐性、或y连锁)一致。可以使用几个参数来定义某人是否受极端数量性状的影响,例如使用最大年龄截止以定义疾患的较早发作,或者数量性状的最小或最大或中位数测量值超出与该性状的正常群体测量值的偏差的定义统计截止(例如,比群体平均值高2个标准偏差)。在一个示例性实施方案中,没有电子健康记录数据的个体可用于该特定数量性状,或者具有关于该特定数量性状的冲突或不可靠数据的个体,无论病历中是否存在该特定数量性状,都可以确定是未知的患病者。
在一些示例性实施方案中,用于生成富集系谱的方法可包括将系谱中的个体识别为患病者或未患病者,其中具有至少一种二元性状、极端数量性状或它们的组合的个体被识别为患病者,并且没有该至少一种二元性状、极端数量性状或它们的组合的个体被识别为未患病者。二元性状可为如上所述定义的ICD代码。几个参数可用于定义如上所述的极端数量性状。在一个示例性实施方案中,没有电子健康记录数据的个体可用于该特定二元性状、数量性状或它们的组合,或者具有关于该特定二元性状、数量性状或它们的组合的冲突或不可靠数据的个体,无论病历中是否存在该特定的数量性状,都可以确定是未知的患病者。
在一些示例性实施方案中,用于生成富集系谱的方法可包括将系谱中的个体识别为患病者或未患病者,其中具有至少一种二元性状、极端数量性状或它们的组合的个体被识别为患病者,并且不具有该至少一种二元性状、极端数量性状或它们的组合的个体被识别为未患病者,并且其中该至少一种二元性状、极端数量性状或它们的组合可包括两个或更多个相似或互补的性状。
在一些示例性实施方案中,用于生成富集系谱的方法可包括将系谱中的个体识别为患病者或未患病者,其中具有至少一种二元性状、极端数量性状或它们的组合的个体被识别为患病者,并且不具有该至少一种二元性状、极端数量性状或它们的组合的个体被识别为未患病者,并且其中该至少一种二元性状、极端数量性状或它们的组合可包括取两个或更多个极端或令人感兴趣的性状的交集。
在一些示例性实施方案中,用于生成富集系谱的方法可包括将系谱中的个体识别为患病者,其中具有至少一种二元性状、极端数量性状或它们的组合的个体被识别为患病者;以及将被确定是患病者的个体定义为来自外部分析的关联结果的患病携带者。
在一些示例性实施方案中,用于生成富集系谱的方法包括基于同期群的测序数据生成个体的一级网络。测序数据可以包括全基因组测序数据、外显子组测序数据或基因型数据。
在一些示例性实施方案中,用于生成富集系谱的方法包括基于外显子组测序数据生成个体的一级网络。可以通过利用群体的相关性来生成基于外显子组测序数据的个体的一级网络,该步骤包括:从自多个人类受试者获得的核酸序列样本数据集中去除低质量的序列变异;为所述样本中的一个或多个样本中的每一样本人建立祖先超类指定;从所述数据集中去除低质量样本;生成祖先超类内的受试者的第一按血统身份估计;独立于受试者的祖先超类生成所述受试者的第二按血统身份估计;以及基于所述第二按血统身份估计中的一个或多个第二按血统身份估计将受试者归类为主要一级家族网络。
在一些示例性实施方案中,用于生成富集系谱的方法包括基于同期群的测序数据生成个体的一级网络,其中所述同期群可包括任何包括多个受试者的数据集。
在一些示例性实施方案中,用于创建富集系谱的方法还包括基于p值来富集该系谱。富集可包括将系谱的“首建者锚定分支”或“分支”定义为系谱内首建者的所有后代,以及使用二项式检验来评估分支是否为针对二元性状富集的。可以使用如上所述的ICD来定义二元性状。富集还可以包括将系谱的“首建者锚定分支”或“分支”定义为系谱中首建者的所有后代,并使用t检验来评估分支是否为针对极端数量性状富集的。几个参数可用于定义如上所述的极端数量性状。此外,富集还可包括应用多重测试p值截止值。
在一个示例性方面中,本公开提供了用于通过以下方式来识别致病变异的方法:通过以下方式生成富集系谱:基于同期群的测序数据生成个体的一级网络;将同期群中的个体识别为患病者或未患病者;创建至少一个包含患病者和未患病者的富集系谱;执行隔离分析以识别在至少一个富集系谱之内和之间共隔离的变异性状对;以及分析该变异性状对以识别所述致病变异。
在一些示例性实施方案中,用于识别致病变异的方法可包括将系谱中的个体识别为患病者或未患病者,其中将具有至少一种二元性状的个体识别为患病者,并且将不具有所述至少一种二元性状的个体识别为未患病者,然后评估所述未患病和未患病个体的模式是否与孟德尔遗传模式(例如,常染色体显性遗传、常染色体隐性遗传、x连锁显性、x连锁隐性、或y连锁)一致。在一些特别的示例性实施方案中,可以使用国际疾病和相关健康问题统计分类(ICD)定义所述二元性状,ICD是由世界卫生组织(WHO)提供的医学分类列表,其包含疾病的代码、体征和症状、异常发现、主诉、社会环境以及伤害或疾病的外部原因。ICD的第九版本或第十版本可用于定义二元性状。在一个示例性实施方案中,没有电子健康记录数据的个体可用于该特定二元性状,或者具有关于该特定二元性状的冲突或不可靠数据的个体,无论病历中是否存在该特定二元性状,都可以确定是未知的患病者。
在一些示例性实施方案中,用于识别致病变异的方法可包括将系谱中的个体识别为患病者或未患病者,其中将具有至少一种极端数量性状的个体识别为患病者,并且将不具有所述至少一种极端数量性状的个体识别为未患病者,然后评估所述未患病和未患病个体的模式是否与孟德尔遗传模式(例如,常染色体显性遗传、常染色体隐性遗传、x连锁显性、x连锁隐性、或y连锁)一致。可以使用几个参数来定义某人是否受极端数量性状的影响,例如使用最大年龄截止以定义疾患的较早发作,或者数量性状的最小或最大或中位数测量值超出与该性状的正常群体测量值的偏差的定义统计截止(例如,比群体平均值高2个标准偏差)。在一个示例性实施方案中,没有电子健康记录数据的个体可用于该特定数量性状,或者具有关于该特定数量性状的冲突或不可靠数据的个体,无论病历中是否存在该特定数量性状,都可以确定是未知的患病者。
在一些示例性实施方案中,用于识别致病变异的方法可包括将系谱中的个体识别为患病者或未患病者,其中具有至少一种二元性状、极端数量性状或它们的组合的个体被识别为患病者,并且没有该至少一种二元性状、极端数量性状或它们的组合的个体被识别为未患病者。二元性状可为如上所述定义的ICD代码。几个参数可用于定义如上所述的极端数量性状。在一个示例性实施方案中,没有电子健康记录数据的个体可用于该特定二元性状、数量性状或它们的组合,或者具有关于该特定二元性状、数量性状或它们的组合的冲突或不可靠数据的个体,无论病历中是否存在该特定的数量性状,都可以确定是未知的患病者。
在一些示例性实施方案中,用于识别致病变异的方法可包括将系谱中的个体识别为患病者或未患病者,其中具有至少一种二元性状、极端数量性状或它们的组合的个体被识别为患病者,并且不具有该至少一种二元性状、极端数量性状或它们的组合的个体被识别为未患病者,并且其中该至少一种二元性状、极端数量性状或它们的组合可包括两个或更多个相似或互补的性状。
在一些示例性实施方案中,用于识别致病变异的方法可包括将系谱中的个体识别为患病者或未患病者,其中具有至少一种二元性状、极端数量性状或它们的组合的个体被识别为患病者,并且不具有该至少一种二元性状、极端数量性状或它们的组合的个体被识别为未患病者,并且其中该至少一种二元性状、极端数量性状或它们的组合可包括取两个或更多个极端或令人感兴趣的性状的交集。
在一些示例性实施方案中,用于识别致病变异的方法可包括将系谱中的个体识别为患病者,其中具有至少一种二元性状、极端数量性状或它们的组合的个体被识别为患病者;以及将被确定是患病者的个体定义为来自外部分析的关联结果的患病携带者。
在一些示例性实施方案中,用于识别致病变异的方法包括基于同期群的测序数据生成个体的一级网络。测序数据可以包括全基因组测序数据、外显子组测序数据或基因型数据。
在一些示例性实施方案中,用于识别致病变异的方法包括基于外显子组测序数据生成个体的一级网络。可以通过利用群体的相关性来生成基于外显子组测序数据的个体的一级网络,该步骤包括:从自多个人类受试者获得的核酸序列样本数据集中去除低质量的序列变异;为所述样本中的一个或多个样本中的每一样本人建立祖先超类指定;从所述数据集中去除低质量样本;生成祖先超类内的受试者的第一按血统身份估计;独立于受试者的祖先超类生成所述受试者的第二按血统身份估计;以及基于所述第二按血统身份估计中的一个或多个第二按血统身份估计将受试者归类为主要一级家族网络。
在一些示例性实施方案中,用于识别致病变异的方法包括基于同期群的测序数据生成个体的一级网络,其中所述同期群可包括任何包括多个受试者的数据集。
在一些示例性实施方案中,用于创建富集系谱的方法还包括基于p值来富集该系谱。富集可包括将系谱的“首建者锚定分支”或“分支”定义为系谱内首建者的所有后代,以及使用二项式检验来评估分支是否为针对二元性状富集的。可以使用如上所述的ICD来定义二元性状。富集还可以包括将系谱的“首建者锚定分支”或“分支”定义为系谱中首建者的所有后代,并使用t检验来评估分支是否为针对极端数量性状富集的。几个参数可用于定义如上所述的极端数量性状。此外,富集还可包括应用多重测试p值截止值。
在一些示例性实施方案中,用于识别致病变异的方法可包括识别与系谱内的患病者共隔离的变异性状对,以及执行隔离分析,该隔离分析包括基于表型隔离找到至少一个富集系谱。隔离可以包括显性隔离模型和隐性隔离模型。在一个示例性实施方案中,基于显性和加性隔离模型找到至少一个富集系谱包括选择具有一种可能的结构的系谱和具有共同祖先的至少三个患病者。该步骤还可包括选择至少一个具有一个或多个相关未患病者的富集系谱以减少假阳性。在另一示例性实施方案中,基于隐性隔离模型找到至少一个富集系谱包括选择具有一种可能结构和多于一个具有未患病亲本的患病者的系谱。该步骤还可包括选择至少一个具有至少两个患病同胞的富集系谱以减少假阳性。
在一些示例性实施方案中,用于识别致病变异的方法包括执行隔离分析以形成特定遗传隔离模型。特定遗传隔离模型可包括显性遗传隔离模型或隐性遗传隔离模型。此外,特定遗传隔离模型还可包括基于其他遗传模式的遗传隔离模型,诸如Y连锁、多因素或线粒体连锁遗传模式。在一个示例性实施方案中,用于识别致病变异的方法包括执行隔离分析以形成显性遗传隔离模型,其中致病变异与患病者针对至少一种二元性状、极端数量性状或它们的组合隔离。在一个示例性实施方案中,用于识别致病变异的方法包括执行隔离分析以形成隐性遗传隔离模型,其中致病变异与为给定基因中的双等位基因变异携带者的患病者隔离,并且如果遗传数据可用于亲本,则所述亲本必须是对于所识别的致病变异为杂合的。
在一些示例性实施方案中,用于识别致病变异的方法可包括执行隔离分析以识别在至少一个富集系谱之内和之间共隔离的变异性状对。在一个示例性实施方案中,用于识别致病变异的方法包括进行隔离分析以识别在多个富集系谱之内和之间共隔离的变异性状对。
在一些示例性实施方案中,用于识别致病变异的方法可包括执行隔离分析以针对家族结构中未包括的感兴趣的表型识别其他患病者的隔离变异或基因。
在一些示例性实施方案中,用于识别致病变异的方法可包括执行隔离分析,所述隔离分析包括将变异和性状与来自群体规模分析的关联结果进行互相参照(crossreferencing)。
在一些示例性实施方案中,用于识别致病变异的方法可包括执行隔离分析以识别先前已知的因果变异和基因。
在一些示例性实施方案中,用于识别致病变异的方法还可包括按照支持系谱/患病者的数目以及按照候选因果变异和基因的数目来对富集系谱进行优先级排序。
在一些示例性实施方案中,用于识别致病变异的方法可包括分析变异性状对,还包括使用足够的家族数据来识别患病者集合,以保证基于家族的关联分析。
在一些示例性实施方案中,用于识别致病变异的方法可包括分析变异性状对,包括在适当的情况下基于系谱和表型信息执行传递不平衡检验(TDT)或其他分析。
在一些示例性实施方案中,用于识别致病变异的方法可包括用于识别几种生理疾患的致病变异的方法。
在一个示例性方面,本公开提供了一种非暂时性计算机可读介质,该非暂时性计算机可读介质存储用于使处理器执行用于生成富集系谱的方法的指令,所述方法包括基于同期群的外显子组测序数据生成个体的一级网络;将该一级网络中的个体识别为患病者或未患病者;以及生成至少一个包含个体的富集系谱,所述至少一个富集系谱包括指定为患病者或未患病者。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于生成富集系谱的方法的指令,所述方法包括:识别系谱中的个体为患病者还是未患病者,其中具有至少一种二元性状的个体被识别为患病者并且没有该至少一种二元性状的个体被识别为未患病者;然后评估患病和未患病的个体的模式是否与孟德尔遗传模式(例如常染色体显性遗传、常染色体隐性遗传、x连锁显性、x连锁隐性、或y连锁)一致。在一些特别的示例性实施方案中,可以使用国际疾病和相关健康问题统计分类(ICD)定义所述二元性状,ICD是由世界卫生组织(WHO)提供的医学分类列表,其包含疾病的代码、体征和症状、异常发现、主诉、社会环境以及伤害或疾病的外部原因。ICD的第九版本或第十版本可用于定义二元性状。在一个示例性实施方案中,没有电子健康记录数据的个体可用于该特定二元性状,或者具有关于该特定二元性状的冲突或不可靠数据的个体,无论病历中是否存在该特定二元性状,都可以确定是未知的患病者。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于生成富集系谱的方法的指令,所述方法包括识别系谱中的个体为患病者还是未患病者,其中具有至少一种极端数量性状的个体被识别为患病者并且没有该至少一种极端数量性状的个体被识别未患病者,然后评估患病和未患病的个体的模式是否与孟德尔遗传模式(例如,常染色体显性遗传、常染色体隐性遗传、x连锁显性、x连锁隐性、或y连锁)一致。可以使用几个参数来定义某人是否受极端数量性状的影响,例如使用最大年龄截止以定义疾患的较早发作,或者数量性状的最小或最大或中位数测量值超出与该性状的正常群体测量值的偏差的定义统计截止(例如,比群体平均值高2个标准偏差)。在一个示例性实施方案中,没有电子健康记录数据的个体可用于该特定数量性状,或者具有关于该特定数量性状的冲突或不可靠数据的个体,无论病历中是否存在该特定数量性状,都可以确定是未知的患病者。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于生成富集系谱的方法的指令,所述方法包括识别系谱中的个体为患病者还是未患病者,其中具有至少一种二元性状、极端数量性状或它们的组合的个体被识别为患病者,并且没有该至少一种二元性状、极端数量性状或它们的组合的个体被识别为未患病者。二元性状可为如上所述定义的ICD代码。几个参数可用于定义如上所述的极端数量性状。在一个示例性实施方案中,没有电子健康记录数据的个体可用于该特定二元性状、数量性状或它们的组合,或者具有关于该特定二元性状、数量性状或它们的组合的冲突或不可靠数据的个体,无论病历中是否存在该特定的数量性状,都可以确定是未知的患病者。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于生成富集系谱的方法的指令,所述方法包括:识别系谱中的个体为患病者还是未患病者,其中具有至少一种二元性状、极端数量性状或它们的组合的个体被识别为患病者,并且没有该至少一种二元性状、极端数量性状或它们的组合的个体被识别为未患病者,并且其中该至少一种二元性状、极端数量性状或它们的组合可包括两个或更多个相似或互补的性状。
在一些示例性实施方案中,非暂时性计算机可读介质存储用于使处理器执行用于生成富集系谱的方法的指令,所述方法包括:识别系谱中的个体为患病者还是未患病者,其中具有至少一种二元性状、极端数量性状或它们的组合的个体被识别为患病者,并且没有该至少一种二元性状、极端数量性状或它们的组合的个体被识别为未患病者,并且其中该至少一种二元性状、极端数量性状或它们的组合可包括取两个或更多个极端或令人感兴趣的性状的交集。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于生成富集系谱的方法的指令,所述方法还可包括:如果同期群中的个体具有至少一种二元性状、极端数量性状或它们的组合,则将该个体识别为患病者;以及将被确定是患病者的个体定义为来自外部分析的关联结果的患病携带者。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于生成富集的系谱的方法的指令,所述方法包括基于同期群的测序数据生成个体的一级网络。测序数据可以包括全基因组测序数据、外显子组测序数据或基因型数据。
在一些示例性实施方案中,非暂时性计算机可读介质存储指令,对的指令用于使处理器执行用于基于外显子组测序数据生成富集系谱的方法。可以通过利用群体的相关性来生成基于外显子组测序数据的个体的一级网络,该步骤包括:从自多个人类受试者获得的核酸序列样本数据集中去除低质量的序列变异;为所述样本中的一个或多个样本中的每一样本人建立祖先超类指定;从所述数据集中去除低质量样本;生成祖先超类内的受试者的第一按血统身份估计;独立于受试者的祖先超类生成所述受试者的第二按血统身份估计;以及基于所述第二按血统身份估计中的一个或多个第二按血统身份估计将受试者归类为主要一级家族网络。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于生成富集系谱的方法的指令,所述方法可包括基于同期群的测序数据生成个体的一级网络,其中该同期群可包括任何包括多个受试者的数据集。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于生成富集系谱的方法的指令,所述方法还可包括基于p值来富集系谱。富集可包括将系谱的“首建者锚定分支”或“分支”定义为系谱内首建者的所有后代,以及使用二项式检验来评估分支是否为针对二元性状富集的。可以使用如上所述的ICD来定义二元性状。富集还可以包括将系谱的“首建者锚定分支”或“分支”定义为系谱中首建者的所有后代,并使用t检验来评估分支是否为针对极端数量性状富集的。几个参数可用于定义如上所述的极端数量性状。此外,富集还可包括应用多重测试p值截止值。
在一个示例性方面中,本公开提供了一种非暂时性计算机可读介质,所述非暂时性计算机可读介质存储用于使处理器执行用于识别致病变异的方法的指令,所述方法包括基于同期群的外显子组测序数据生成个体的一级网络;将该一级网络中的个体识别为患病者或未患病者;创建包含所述个体的至少一个富集系谱,包括指定为患病者或未患病者;执行隔离分析以识别在至少一个富集系谱之内和之间共隔离的变异性状对;以及分析所述变异性状对以确定所述致病变异。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于识别致病变异的方法的指令,所述方法包括:识别系谱中的个体为患病者还是未患病者,其中具有至少一种二元性状的个体被识别为患病者并且没有该至少一种二元性状的个体被识别为未患病者;然后评估患病和未患病的个体的模式是否与孟德尔遗传模式(例如常染色体显性遗传、常染色体隐性遗传、x连锁显性、x连锁隐性、或y连锁)一致。在一些特别的示例性实施方案中,可以使用国际疾病和相关健康问题统计分类(ICD)定义所述二元性状,ICD是由世界卫生组织(WHO)提供的医学分类列表,其包含疾病的代码、体征和症状、异常发现、主诉、社会环境以及伤害或疾病的外部原因。ICD的第九版本或第十版本可用于定义二元性状。在一个示例性实施方案中,没有电子健康记录数据的个体可用于该特定二元性状,或者具有关于该特定二元性状的冲突或不可靠数据的个体,无论病历中是否存在该特定二元性状,都可以确定是未知的患病者。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于识别致病变异的方法的指令,所述方法包括识别系谱中的个体为患病者还是未患病者,其中具有至少一种极端数量性状的个体被识别为患病者并且没有该至少一种极端数量性状的个体被识别未患病者,然后评估患病和未患病的个体的模式是否与孟德尔遗传模式(例如,常染色体显性遗传、常染色体隐性遗传、x连锁显性、x连锁隐性、或y连锁)一致。可以使用几个参数来定义某人是否受极端数量性状的影响,例如使用最大年龄截止以定义疾患的较早发作,或者数量性状的最小或最大或中位数测量值超出与该性状的正常群体测量值的偏差的定义统计截止(例如,比群体平均值高2个标准偏差)。在一个示例性实施方案中,没有电子健康记录数据的个体可用于该特定数量性状,或者具有关于该特定数量性状的冲突或不可靠数据的个体,无论病历中是否存在该特定数量性状,都可以确定是未知的患病者。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于识别致病变异的方法的指令,所述方法包括识别系谱中的个体为患病者还是未患病者,其中具有至少一种二元性状、极端数量性状或它们的组合的个体被识别为患病者,并且没有该至少一种二元性状、极端数量性状或它们的组合的个体被识别为未患病者。二元性状可为如上所述定义的ICD代码。几个参数可用于定义如上所述的极端数量性状。在一个示例性实施方案中,没有电子健康记录数据的个体可用于该特定二元性状、数量性状或它们的组合,或者具有关于该特定二元性状、数量性状或它们的组合的冲突或不可靠数据的个体,无论病历中是否存在该特定的数量性状,都可以确定是未知的患病者。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于识别致病变异的方法的指令,所述方法包括:识别系谱中的个体为患病者还是未患病者,其中具有至少一种二元性状、极端数量性状或它们的组合的个体被识别为患病者,并且没有该至少一种二元性状、极端数量性状或它们的组合的个体被识别为未患病者,并且其中该至少一种二元性状、极端数量性状或它们的组合可包括两个或更多个相似或互补的性状。
在一些示例性实施方案中,非暂时性计算机可读介质存储用于使处理器执行用于识别致病变异的方法的指令,所述方法包括:识别系谱中的个体为患病者还是未患病者,其中具有至少一种二元性状、极端数量性状或它们的组合的个体被识别为患病者,并且没有该至少一种二元性状、极端数量性状或它们的组合的个体被识别为未患病者,并且其中该至少一种二元性状、极端数量性状或它们的组合可包括取两个或更多个极端或令人感兴趣的性状的交集。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于识别致病变异的方法的指令,所述方法还可包括:如果同期群中的个体具有至少一种二元性状、极端数量性状或它们的组合,则将该个体识别为患病者;以及将被确定是患病者的个体定义为来自外部分析的关联结果的患病携带者。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于识别致病变异的方法的指令,所述方法包括基于同期群的测序数据生成个体的一级网络。测序数据可以包括全基因组测序数据、外显子组测序数据或基因型数据。
在一些示例性实施方案中,非暂时性计算机可读介质存储用于使处理器执行用于基于外显子组测序数据识别致病变异的方法的指令。可以通过利用群体的相关性来生成基于外显子组测序数据的个体的一级网络,该步骤包括:从自多个人类受试者获得的核酸序列样本数据集中去除低质量的序列变异;为所述样本中的一个或多个样本中的每一样本人建立祖先超类指定;从所述数据集中去除低质量样本;生成祖先超类内的受试者的第一按血统身份估计;独立于受试者的祖先超类生成所述受试者的第二按血统身份估计;以及基于所述第二按血统身份估计中的一个或多个第二按血统身份估计将受试者归类为主要一级家族网络。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于识别致病变异的方法的指令,所述方法可包括基于同期群的测序数据生成个体的一级网络,其中该同期群可包括任何包括多个受试者的数据集。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于识别致病变异的方法的指令,所述方法还可包括基于p值来富集系谱。富集可包括将系谱的“首建者锚定分支”或“分支”定义为系谱内首建者的所有后代,以及使用二项式检验来评估分支是否为针对二元性状富集的。可以使用如上所述的ICD来定义二元性状。富集还可以包括将系谱的“首建者锚定分支”或“分支”定义为系谱中首建者的所有后代,并使用t检验来评估分支是否为针对极端数量性状富集的。几个参数可用于定义如上所述的极端数量性状。此外,富集还可包括应用多重测试p值截止
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于识别致病变异的方法的指令,所述方法可包括识别与系谱内的患病者共隔离的变异性状对,以及执行隔离分析,该隔离分析包括基于表型隔离找到至少一个富集系谱。隔离可以包括显性隔离模型和隐性隔离模型。在一个示例性实施方案中,基于显性和加性隔离模型找到至少一个富集系谱包括选择具有一种可能的结构的系谱和具有共同祖先的至少三个患病者。该步骤还可包括选择至少一个具有一个或多个相关未患病者的富集系谱以减少假阳性。在另一示例性实施方案中,基于隐性隔离模型找到至少一个富集系谱包括选择具有一种可能结构和多于一个具有未患病亲本的患病者的系谱。该步骤还可包括选择至少一个具有至少两个患病同胞的富集系谱以减少假阳性。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于识别致病变异的方法的指令,所述方法可包括执行隔离分析以形成特定遗传隔离模型。特定遗传隔离模型可包括显性遗传隔离模型或隐性遗传隔离模型。此外,特定遗传隔离模型还可包括基于其他遗传模式的遗传隔离模型,诸如Y连锁、多因素或线粒体连锁遗传模式。在一个示例性实施方案中,用于识别致病变异的方法包括执行隔离分析以形成显性遗传隔离模型,其中致病变异与患病者针对至少一种二元性状、极端数量性状或它们的组合隔离。在一个示例性实施方案中,用于识别致病变异的方法包括执行隔离分析以形成隐性遗传隔离模型,其中致病变异与为给定基因中的双等位基因变异携带者的患病者隔离,并且如果遗传数据可用于亲本,则所述亲本必须是对于所识别的致病变异为杂合的。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于识别致病变异的方法的指令,所述方法可包括执行隔离分析以识别在至少一个富集系谱之内和之间共隔离的变异性状对。在一个示例性实施方案中,用于识别致病变异的方法包括进行隔离分析以识别在多个富集系谱之内和之间共隔离的变异性状对。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于识别致病变异的方法的指令,所述方法可包括执行隔离分析以针对家族结构中未包括的感兴趣的表型识别其他患病者的隔离变异或基因。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于识别致病变异的方法的指令,所述方法可包括执行隔离分析,所述隔离分析包括将变异和性状与来自群体规模分析的关联结果进行相互参照。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于识别致病变异的方法的指令,所述方法可包括执行隔离分析以识别先前已知的因果变异和基因。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于识别致病变异的方法的指令,所述方法可包括按照支持系谱/患病者的数目以及按照候选因果变异和基因的数目来对富集系谱进行优先级排序。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于识别致病变异的方法的指令,所述方法可包括分析变异性状对,还包括使用足够的家族数据来识别患病者集合,以保证基于家族的关联分析。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于识别致病变异的方法的指令,所述方法可包括分析变异性状对,包括在适当的情况下基于系谱和表型信息执行传递不平衡检验(TDT)或其他分析。
在一些示例性实施方案中,所述非暂时性计算机可读介质存储用于使处理器执行用于识别几种生理疾患的致病变异的方法的指令。
在一个示例性方面中,本公开提供一种用于生成富集系谱的系统,所述系统包括数据处理器和与该数据处理器耦合的存储器,所述处理器被配置为基于同期群的排序数据生成个体的一级网络;识别该一级网络中的个体为患病者还是未患病者;以及生成包含所述个体的至少一个富集系谱,包括指定为患病者或未患病者。
在一些示例性实施方案中,所述用于生成富集系谱的系统包括数据处理器和与数据处理器耦合的存储器,所述处理器被配置为识别系谱中的个体为患病者还是未患病者,其中具有至少一种二元性状的个体被识别为患病者并且没有该至少一种二元性状的个体被识别为未患病者;然后评估患病和未患病的个体的模式是否与孟德尔遗传模式(例如常染色体显性遗传、常染色体隐性遗传、x连锁显性、x连锁隐性、或y连锁)一致。在一些特别的示例性实施方案中,可以使用国际疾病和相关健康问题统计分类(ICD)定义所述二元性状,ICD是由世界卫生组织(WHO)提供的医学分类列表,其包含疾病的代码、体征和症状、异常发现、主诉、社会环境以及伤害或疾病的外部原因。ICD的第九版本或第十版本可用于定义二元性状。在一个示例性实施方案中,没有电子健康记录数据的个体可用于该特定二元性状,或者具有关于该特定二元性状的冲突或不可靠数据的个体,无论病历中是否存在该特定二元性状,都可以确定是未知的患病者。
在一些示例性实施方案中,所述用于生成富集系谱的系统包括数据处理器和与数据处理器耦合的存储器,所述处理器被配置为识别系谱中的个体为患病者还是未患病者,其中具有至少一种极端数量性状的个体被识别为患病者并且没有该至少一种极端数量性状的个体被识别未患病者,然后评估患病和未患病的个体的模式是否与孟德尔遗传模式(例如,常染色体显性遗传、常染色体隐性遗传、x连锁显性、x连锁隐性、或y连锁)一致。可以使用几个参数来定义某人是否受极端数量性状的影响,例如使用最大年龄截止以定义疾患的较早发作,或者数量性状的最小或最大或中位数测量值超出与该性状的正常群体测量值的偏差的定义统计截止(例如,比群体平均值高2个标准偏差)。在一个示例性实施方案中,没有电子健康记录数据的个体可用于该特定数量性状,或者具有关于该特定数量性状的冲突或不可靠数据的个体,无论病历中是否存在该特定数量性状,都可以确定是未知的患病者。
在一些示例性实施方案中,所述用于生成富集系谱的系统包括数据处理器和与数据处理器耦合的存储器,所述处理器被配置为识别系谱中的个体为患病者还是未患病者,其中具有至少一种二元性状、极端数量性状或它们的组合的个体被识别为患病者,并且没有该至少一种二元性状、极端数量性状或它们的组合的个体被识别为未患病者。二元性状可为如上所述定义的ICD代码。几个参数可用于定义如上所述的极端数量性状。在一个示例性实施方案中,没有电子健康记录数据的个体可用于该特定二元性状、数量性状或它们的组合,或者具有关于该特定二元性状、数量性状或它们的组合的冲突或不可靠数据的个体,无论病历中是否存在该特定的数量性状,都可以确定是未知的患病者。
在一些示例性实施方案中,所述用于生成富集系谱的系统包括数据处理器和与数据处理器耦合的存储器,所述处理器被配置为识别系谱中的个体为患病者还是未患病者,其中具有至少一种二元性状、极端数量性状或它们的组合的个体被识别为患病者,并且没有该至少一种二元性状、极端数量性状或它们的组合的个体被识别为未患病者,并且其中该至少一种二元性状、极端数量性状或它们的组合可包括两个或更多个相似或互补的性状。
在一些示例性实施方案中,所述用于生成富集系谱的系统包括数据处理器和与所述数据处理器耦合的存储器,所述处理器被配置为识别系谱中的个体为患病者还是未患病者,其中具有至少一种二元性状、极端数量性状或它们的组合的个体被识别为患病者,并且没有该至少一种二元性状、极端数量性状或它们的组合的个体被识别为未患病者,并且其中该至少一种二元性状、极端数量性状或它们的组合可包括取两个或更多个极端或令人感兴趣的性状的交集。
在一些示例性实施方案中,所述用于生成富集系谱的系统包括数据处理器和与所述数据处理器耦合的存储器,所述处理器被配置为如果同期群中的个体具有至少一种二元性状、极端数量性状或它们的组合,则将该个体识别为患病者;以及将被确定是患病者的个体定义为来自外部分析的关联结果的患病携带者。
在一些示例性实施方案中,所述用于生成富集系谱的系统包括数据处理器和与所述数据处理器耦合的存储器,所述处理器被配置为基于同期群的排序数据生成个体的一级网络。测序数据可以包括全基因组测序数据、外显子组测序数据或基因型数据。
在一些示例性实施方案中,所述用于生成富集系谱的系统包括数据处理器和与所述数据处理器耦合的存储器,所述处理器被配置为基于外显子组测序数据生成个体的一级网络。可以通过利用群体的相关性来生成基于外显子组测序数据的个体的一级网络,该步骤包括:从自多个人类受试者获得的核酸序列样本数据集中去除低质量的序列变异;为所述样本中的一个或多个样本中的每一样本人建立祖先超类指定;从所述数据集中去除低质量样本;生成祖先超类内的受试者的第一按血统身份估计;独立于受试者的祖先超类生成所述受试者的第二按血统身份估计;以及基于所述第二按血统身份估计中的一个或多个第二按血统身份估计将受试者归类为主要一级家族网络。
在一些示例性实施方案中,所述用于生成富集系谱的系统包括数据处理器和与所述数据处理器耦合的存储器,所述处理器被配置为基于同期群的排序数据生成个体的一级网络,其中该同期群可包括任何包含多个受试者的数据集。
在一些示例性实施方案中,所述用于生成富集系谱的系统包括数据处理器和与所述数据处理器耦合的存储器,所述处理器被配置为还包括基于p值来富集系谱。富集可包括将系谱的“首建者锚定分支”或“分支”定义为系谱内首建者的所有后代,以及使用二项式检验来评估分支是否为针对二元性状富集的。可以使用如上所述的ICD来定义二元性状。富集还可以包括将系谱的“首建者锚定分支”或“分支”定义为系谱中首建者的所有后代,并使用t检验来评估分支是否为针对极端数量性状富集的。几个参数可用于定义如上所述的极端数量性状。此外,富集还可包括应用多重测试p值截止值。
在一个示例性方面中,本公开提供了一种用于识别致病变异的系统,所述系统包括数据处理器和与该数据处理器耦合的存储器,所述处理器被配置为基于同期群的排序数据生成个体的一级网络;识别该一级网络中的个体为患病者还是未患病者;以及生成包含所述个体的至少一个富集系谱,包括指定为患病者或未患病者。
在一些示例性实施方案中,所述用于识别致病变异的系统包括数据处理器和与数据处理器耦合的存储器,所述处理器被配置为识别系谱中的个体为患病者还是未患病者,其中具有至少一种二元性状的个体被识别为患病者并且没有该至少一种二元性状的个体被识别为未患病者;然后评估患病和未患病的个体的模式是否与孟德尔遗传模式(例如常染色体显性遗传、常染色体隐性遗传、x连锁显性、x连锁隐性、或y连锁)一致。在一些特别的示例性实施方案中,可以使用国际疾病和相关健康问题统计分类(ICD)定义所述二元性状,ICD是由世界卫生组织(WHO)提供的医学分类列表,其包含疾病的代码、体征和症状、异常发现、主诉、社会环境以及伤害或疾病的外部原因。ICD的第九版本或第十版本可用于定义二元性状。在一个示例性实施方案中,没有电子健康记录数据的个体可用于该特定二元性状,或者具有关于该特定二元性状的冲突或不可靠数据的个体,无论病历中是否存在该特定二元性状,都可以确定是未知的患病者。
在一些示例性实施方案中,所述用于识别致病变异的系统包括数据处理器和与数据处理器耦合的存储器,所述处理器被配置为识别系谱中的个体为患病者还是未患病者,其中具有至少一种极端数量性状的个体被识别为患病者并且没有该至少一种极端数量性状的个体被识别未患病者,然后评估患病和未患病的个体的模式是否与孟德尔遗传模式(例如,常染色体显性遗传、常染色体隐性遗传、x连锁显性、x连锁隐性、或y连锁)一致。可以使用几个参数来定义某人是否受极端数量性状的影响,例如使用最大年龄截止以定义疾患的较早发作,或者数量性状的最小或最大或中位数测量值超出与该性状的正常群体测量值的偏差的定义统计截止(例如,比群体平均值高2个标准偏差)。在一个示例性实施方案中,没有电子健康记录数据的个体可用于该特定数量性状,或者具有关于该特定数量性状的冲突或不可靠数据的个体,无论病历中是否存在该特定数量性状,都可以确定是未知的患病者。
在一些示例性实施方案中,所述用于识别致病变异的系统包括数据处理器和与数据处理器耦合的存储器,所述处理器被配置为识别系谱中的个体为患病者还是未患病者,其中具有至少一种二元性状、极端数量性状或它们的组合的个体被识别为患病者,并且没有该至少一种二元性状、极端数量性状或它们的组合的个体被识别为未患病者。二元性状可为如上所述定义的ICD代码。几个参数可用于定义如上所述的极端数量性状。在一个示例性实施方案中,没有电子健康记录数据的个体可用于该特定二元性状、数量性状或它们的组合,或者具有关于该特定二元性状、数量性状或它们的组合的冲突或不可靠数据的个体,无论病历中是否存在该特定的数量性状,都可以确定是未知的患病者。
在一些示例性实施方案中,所述用于识别致病变异的系统包括数据处理器和与数据处理器耦合的存储器,所述处理器被配置为识别系谱中的个体为患病者还是未患病者,其中具有至少一种二元性状、极端数量性状或它们的组合的个体被识别为患病者,并且没有该至少一种二元性状、极端数量性状或它们的组合的个体被识别为未患病者,并且其中该至少一种二元性状、极端数量性状或它们的组合可包括两个或更多个相似或互补的性状。
在一些示例性实施方案中,所述用于识别致病变异的系统包括数据处理器和与所述数据处理器耦合的存储器,所述处理器被配置为识别系谱中的个体为患病者还是未患病者,其中具有至少一种二元性状、极端数量性状或它们的组合的个体被识别为患病者,并且没有该至少一种二元性状、极端数量性状或它们的组合的个体被识别为未患病者,并且其中该至少一种二元性状、极端数量性状或它们的组合可包括取两个或更多个极端或令人感兴趣的性状的交集。
在一些示例性实施方案中,所述用于识别致病变异的系统包括数据处理器和与所述数据处理器耦合的存储器,所述处理器被配置为如果同期群中的个体具有至少一种二元性状、极端数量性状或它们的组合,则将该个体识别为患病者;以及将被确定是患病者的个体定义为来自外部分析的关联结果的患病携带者。
在一些示例性实施方案中,所述用于识别致病变异的系统包括数据处理器和与所述数据处理器耦合的存储器,所述处理器被配置为基于同期群的排序数据生成个体的一级网络。测序数据可以包括全基因组测序数据、外显子组测序数据或基因型数据。
在一些示例性实施方案中,所述用于识别致病变异的系统包括数据处理器和与所述数据处理器耦合的存储器,所述处理器被配置为基于外显子组测序数据生成个体的一级网络。可以通过利用群体的相关性来生成基于外显子组测序数据的个体的一级网络,该步骤包括:从自多个人类受试者获得的核酸序列样本数据集中去除低质量的序列变异;为所述样本中的一个或多个样本中的每一样本人建立祖先超类指定;从所述数据集中去除低质量样本;生成祖先超类内的受试者的第一按血统身份估计;独立于受试者的祖先超类生成所述受试者的第二按血统身份估计;以及基于所述第二按血统身份估计中的一个或多个第二按血统身份估计将受试者归类为主要一级家族网络。
在一些示例性实施方案中,所述用于识别致病变异的系统包括数据处理器和与所述数据处理器耦合的存储器,所述处理器被配置为基于同期群的排序数据生成个体的一级网络,其中该同期群可包括任何包含多个受试者的数据集。
在一些示例性实施方案中,所述用于识别致病变异的系统包括数据处理器和与所述数据处理器耦合的存储器,所述处理器被配置为还包括基于p值来富集系谱。富集可包括将系谱的“首建者锚定分支”或“分支”定义为系谱内首建者的所有后代,以及使用二项式检验来评估分支是否为针对二元性状富集的。可以使用如上所述的ICD来定义二元性状。富集还可以包括将系谱的“首建者锚定分支”或“分支”定义为系谱中首建者的所有后代,并使用t检验来评估分支是否为针对极端数量性状富集的。几个参数可用于定义如上所述的极端数量性状。此外,富集还可包括应用多重测试p值截止值。
在一些示例性实施方案中,所述用于识别致病变异的系统包括数据处理器和与所述数据处理器耦合的存储器,所述处理器被配置为识别与系谱内的患病者共隔离的变异性状对,以及执行隔离分析,该隔离分析包括基于表型隔离找到至少一个富集系谱。隔离可以包括显性隔离模型和隐性隔离模型。在一个示例性实施方案中,基于显性和加性隔离模型找到至少一个富集系谱包括选择具有一种可能的结构的系谱和具有共同祖先的至少三个患病者。该步骤还可包括选择至少一个具有一个或多个相关未患病者的富集系谱以减少假阳性。在另一示例性实施方案中,基于隐性隔离模型找到至少一个富集系谱包括选择具有一种可能结构和多于一个具有未患病亲本的患病者的系谱。该步骤还可包括选择至少一个具有至少两个患病同胞的富集系谱以减少假阳性。
在一些示例性实施方案中,所述用于识别致病变异的系统包括数据处理器和与该数据处理器耦合的存储器,所述处理器被配置为执行隔离分析以形成特定遗传隔离模型。特定遗传隔离模型可包括显性遗传隔离模型或隐性遗传隔离模型。此外,特定遗传隔离模型还可包括基于其他遗传模式的遗传隔离模型,诸如Y连锁、多因素或线粒体连锁遗传模式。在一个示例性实施方案中,用于识别致病变异的方法包括执行隔离分析以形成显性遗传隔离模型,其中致病变异与患病者针对至少一种二元性状、极端数量性状或它们的组合隔离。在一个示例性实施方案中,用于识别致病变异的方法包括执行隔离分析以形成隐性遗传隔离模型,其中致病变异与为给定基因中的双等位基因变异携带者的患病者隔离,并且如果遗传数据可用于亲本,则所述亲本必须是对于所识别的致病变异为杂合的。
在一些示例性实施方案中,所述用于识别致病变异的系统包括数据处理器和与所述数据处理器耦合的存储器,所述处理器被配置为执行隔离分析以识别在至少一个富集系谱之内和之间共隔离的变异性状对。在一个示例性实施方案中,用于识别致病变异的方法包括进行隔离分析以识别在多个富集系谱之内和之间共隔离的变异性状对。
在一些示例性实施方案中,所述用于识别致病变异的系统包括数据处理器和与所述数据处理器耦合的存储器,所述处理器被配置为执行隔离分析以针对家族结构中未包括的感兴趣的表型识别其他患病者的隔离变异或基因。
在一些示例性实施方案中,所述用于识别致病变异的系统包括数据处理器和与所述数据处理器耦合的存储器,所述处理器被配置为执行隔离分析,所述隔离分析包括将变异和性状与来自群体规模分析的关联结果进行相互参照。
在一些示例性实施方案中,所述用于识别致病变异的系统包括数据处理器和与所述数据处理器耦合的存储器,所述处理器被配置为执行隔离分析以识别先前已知的因果变异和基因。
在一些示例性实施方案中,所述用于识别致病变异的系统包括数据处理器和与所述数据处理器耦合的存储器,所述处理器被配置为按照支持系谱/患病者的数目以及按照候选因果变异和基因的数目来对富集系谱进行优先级排序。
在一些示例性实施方案中,所述用于识别致病变异的系统包括数据处理器和与所述数据处理器耦合的存储器,所述处理器被配置为分析变异性状对,还包括使用足够的家族数据来识别患病者集合,以保证基于家族的关联分析。
在一些示例性实施方案中,所述用于识别致病变异的系统包括数据处理器和与所述数据处理器耦合的存储器,所述处理器被配置为分析变异性状对,包括在适当的情况下基于系谱和表型信息执行传递不平衡检验(TDT)或其他分析。
在一些示例性实施方案中,所述用于识别致病变异的系统包括数据处理器和与所述数据处理器耦合的存储器,所述处理器被配置为识别几种生理疾患的致病变异。
本文所述的方法和系统可以(i)提供对致病分子机制的更好理解,(ii)导致更好的疾病分类和更好的管理,(iii)提供对与相关基因变异有关的差异代谢的鉴定(使用与癌细胞中改变的代谢有关的关键酶或蛋白质或受体作为新药开发的靶标),(iv)为疾病如癌症提供精确的分类预测,这可以帮助预测未来的临床病程和存活期,以及(v)通过识别致病遗传缺陷来设计基因疗法(通过增加所需但不足的基因,或阻断有害基因(通过反义寡核糖核苷酸或转录因子诱饵,或特定的适体))。
附图说明
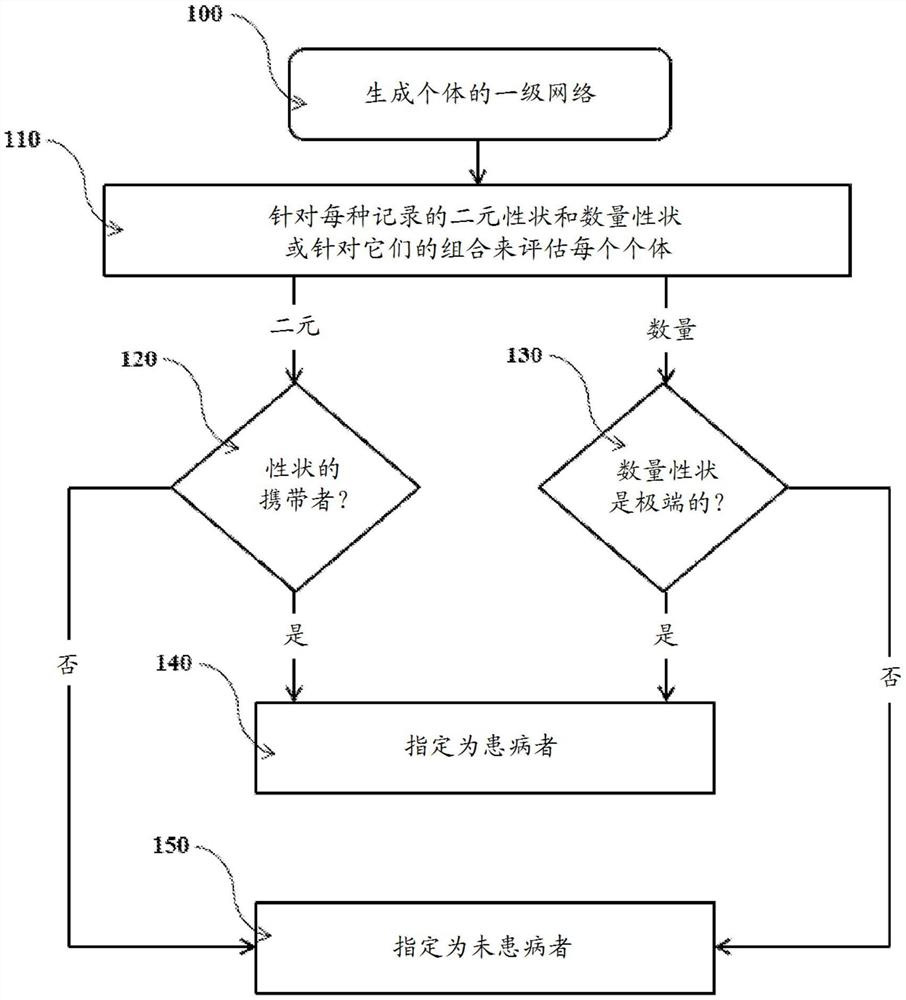
图1是执行系谱富集的本发明示例性实施方案的流程图。
图2是执行系谱富集的本发明示例性实施方案的流程图。
图3是示例性操作环境。
图4示出了被配置用于执行所公开的方法的多个系统部件。
图5示出了根据示例性实施方案确定的来自DiscovEHR同期群的前92K测序个体的IBD0对比IDB1图。
图6示出了来自DiscovEHR同期群的针对原发性血友病表型(Phe10_D685,ICD10CMD68.5)的几个富集系谱,其中根据示例性实施方案执行系谱富集。
图7A和图7B示出了遗传性出血性毛细血管扩张表型的两个富集系谱(Phe10_I780,ICD10CM I78.0),其中根据示例性实施方案执行系谱富集。
图8示出了来自DiscovEHR同期群的系谱,所述系谱包括表现出遗传性出血性毛细血管扩张表型(Phe10_I780,ICD10CM I78.0)的变异隔离的富集系谱,其中根据示例性实施方案执行系谱富集和隔离分析。
图9示出了来自DiscovEHR同期群的针对肺气肿表型的几个富集系谱,其中根据示例性实施方案执行系谱富集。
图10示出了来自DiscovEHR同期群的针对肾移植表型的富集系谱(Phe9_V420,ICD9CM V42.0),其中根据示例性实施方案执行系谱富集。
图11示出了来自DiscovEHR同期群的针对终末期肾脏疾病表型(Phe9_5856,ICD9CM 585.6)的几个富集系谱,其中根据示例性实施方案执行系谱富集。
图12示出了来自DiscovEHR同期群的针对遗传性运动和感觉神经病表型(Charcot-Marie-Tooth疾病)(Phe10_G600,ICD10CM G60.0)表型的富集系谱。
图13是示出在各种组织中编码的原肌球蛋白2(TMP2)基因的每百万转录本(TPM)的基因表达数据的图表
图14示出了来自DiscovEHR同期群的针对双相障碍的富集系谱,其中根据示例性实施方案执行系谱富集和隔离分析。
图15是示出在各种组织中编码的染色体20开放阅读框203(C20orf203)的每百万转录本(TPM)的基因表达数据的图。
图16示出了来自DiscovEHR同期群的针对双相障碍表型的富集系谱,其中根据示例性实施方案执行系谱富集。
图17示出了来自DiscovEHR同期群的针对双相障碍表型的富集系谱,其中根据示例性实施方案执行系谱富集
图18示出了来自DiscovEHR同期群的针对双相障碍表型的富集系谱,其中根据示例性实施方案执行系谱富集
图19是示出了在各种组织中的微头蛋白1(microcephalin 1)(MCPH 1)的每百万转录本(TPM)的基因表达数据的图表。
图20示出了来自DiscovEHR同期群的针对家族性地中海贫血表型的富集系谱,其中根据示例性实施方案执行系谱富集。
图21示出了来自DiscovEHR同期群的针对碱性磷酸酶门诊患者中心趋势值的富集系谱,其中根据示例性实施方案执行系谱富集
具体实施方式
术语“一个(种)”应理解为是指“至少一个(种)”;并且术语“约”和“大致”应被理解为允许如本领域普通技术人员将理解的标准变化;并且在提供范围的地方,包括端点。
基于家族的关联研究使用病例-对照设计,其中病例来自医院或疾病登记处。对照可以是无关的(例如,基于群体或医院/登记簿),也可以是病例的家族成员(例如,亲本或同胞)。比较病例与对照中给定等位基因的出现,以查看基因与疾病之间是否存在“关联”。随着大规模单核苷酸多态性(SNP)基因分型的可用性,关联研究变得越来越普遍,并迅速从所关注候选基因的研究扩展到全基因组关联研究。
下一代测序策略的出现为阐明这些疾病中的遗传缺陷提供了广阔的前景。整个基因组(约30亿个碱基对)目前可以在几天的时段内进行测序,并且成本正在迅速下降,使其可以作为常规研究工具使用。对基因组的蛋白质编码部分进行测序(称为外显子组测序)对于发现致病基因更为有效,因为该外显子组只占基因组的一小部分(约38Mb),并且因为外显子包含孟德尔基因中绝大部分的已知突变(Albert等人,Nature Methods(2007)4:903-905;Gnirke等人,Nature Biotechnology(2009)27:182-189;Hodges等人,NatureGenetics(2007)9:1522-1527;Majewski等人,Journal of Medical Genetics(2011)48:580-589)。因此,外显子组测序非常适合寻找具有可疑遗传原因的疾患中的突变,而无需事先了解候选基因或途径。
大型人类测序研究中的许多大型人类测序研究均从综合医疗保健群体中收集具有伴随的表型丰富的电子健康记录(EHR)的样本,目的是将EHR和基因组序列数据结合起来,以催化转化探索和精准医学。来自这些项目的数据可用于识别性状和疾病的某些遗传驱动因素。
如果病例和对照来自具有不同等位基因频率的不同源群体,从而导致群体分层,则可能检测到假关联(Cardon和Palmer.Lancet(2003)361(9357):598-604)。关于这种混杂可能导致多少偏差存在争议(Wacholder等人,Cancer Epidemiology,Biomarkers&Prevention(2002)11(6):513-520;Thomas and Witte.Cancer Epidemiology,Biomarkers&Prevention(2002)11(6):502-512;Gorroochurn等人,Human Heredity(2004)58(1):40-48)。可以通过使用基于家族的研究设计来避免群体分层。在研究亲本及其后代或同胞时,每个家族内的病例和对照来自同一源群体。常见的基于家族的病例-对照设计是亲本三重奏(例如,传递不平衡检验(TDT)方法)和同胞对照。也可以研究其他亲属(例如,堂兄)或同时研究大量不同的家族成员。
识别大型同期群内的家族涉及识别由足够信息丰富的患病个体组成的系谱,以使给定性状适用于基于家族的遗传研究。当查询家族内与给定的感兴趣表型共隔离的具有潜在中等至大效应的罕见变异时,系谱为特别信息丰富的。可以利用这些系谱来帮助定义具有感兴趣表型的相关参与者的子集,然后检查这些子集以识别性状和疾病的遗传驱动因素。
本公开至少部分地基于以下认识:关于多个受试者的基因组样本的数据集内的个体的一级网络的信息尤其允许研究罕见遗传变异与疾病之间的联系。
本文所述的方法可以应用于各种类型的基因组样本数据集。数据集类型的非限制性示例包括单医疗保健网络群体;多医疗保健网络群体;在种族、文化或社会方面同质或异质的群体;混合年龄群体或在年龄方面同质的群体;地理上集中或分散的群体;或它们的组合。该数据集可以具有各种类型的遗传变异。可评定的遗传变异类型的非限制性示例包括点突变、插入、缺失、倒位、重复和多聚化。可以获取遗传变异的手段的非限制性示例包括以下步骤:
-样品制备和测序(Dewey等人(2016),Science 354,aaf6814-1至aaf6814-10);
-在测序完成时,可以来自每次测序运行的原始数据收集到本地缓冲存储器中,并上传到DNAnexus平台(Reid等人(2014);BMC Bioinformatics 15,30)以进行自动化分析。
-可以使用CASAVA(Illumina Inc.,San Diego,CA)生成样本级读取文件,并使用BWA-mem(Li和Durbin(2009);Bioinformatics 25,1754-176;Li(2013);arXiv q-bio.GN)将其与GRCh38比对。
-可以使用GATK(McKenna等人(2010);Genome Res.20,1297-1303)和Picard处理所得的BAM文件,以对推定的插入缺失附近的读段进行分选、标记重复和执行局部重排。
-可以使用Ensembl85基因定义,用snpEFF(Cingolani等人(2012);Fly(Austin)6,80-92)对已测序的变体进行注释,以确定对转录本和基因的功能影响。
本文所述的方法可以用于识别引起生理疾患的致病变异。非限制性示例包括心理疾患、血液相关疾患、疼痛相关疾患、激素相关疾患、肺部疾病、牙齿疾患、生育相关疾患、精神疾患、运动疾患、心血管疾患、循环系统疾患、自身免疫性疾病、炎性疾病、肾疾患、肝疾患、遗传性出血性毛细血管扩张、运动感觉神经病、家族性主动脉瘤、甲状腺癌、色素性青光眼、家族性高胆固醇血症,或它们的组合。
应当理解的是所述方法不限于上述步骤中的任何步骤,并且可以通过任何合适的手段进行序列变体的获取。
本公开还至少部分地基于以下认识:从关于多个受试者的基因组样本数据集内的一级亲属的信息生成的系谱可以提供信息,以识别家族中隔离的罕见变异。
已经开发了几种可用于识别一级亲属的统计方法。一个此类非限制性示例是通过计算个体的按血统身份(IBD)估计以识别数据集内的不同类型的家族关系,并且可以使用PRIMUS(Staples等人,(2014),Am.J.Hum.Genet.95,553-564)将成对的关系分类为不同的家族类别并重建系谱。仅应包括数据集之间的估计的一级关系。例如,为了从包含外显子组测序数据的数据集中识别一级亲属,可以利用如在于2018年9月7日提交的发明名称为“SYSTEMS AND METHODS FOR LEVERAGING RELATEDNESS IN GENOMIC DATA ANALYSIS”的共同待决美国专利公开号20190205502中描述的方法,该共同待决美国专利公开据此以引用方式整体并入。
为了从多个受试者的基因组样本的数据集生成系谱,可以使用几种方法,诸如COP(构建远交系谱)和CIP(构建近交系谱)、IPED(基于遗传路径的系谱重建)和IPED2、PREPARE(亲属划分),以及最大无关集的系谱重建和识别(PRIMUS)(Riester等人,Bioinformatics(2009)25:2134-2139;Hadfield等人,Molecular Ecology(2006)15:3715-3730;Marshall等人,Molecular Ecology(1998)7:639-655;Cussens等人,Genetic Epidemiology(2013)37:69-83;He等人,Journal of Computational Biology(2013)20:780-792;Kirkpatrick等人,Journal of Computational Biology(2011)18:1481-1493;Staples等人,GeneticEpidemiology(2013)37:136-141;Shem-Tov and Halperin.PLoS Computational Biology(2014)10:e1003610)。也可以使用其他方法,诸如PLINK、KING和KINSHIP。
应当理解的是,本公开不限于上述数据集、识别一级亲属和/或生成系谱的方法中的任一者,并且可以通过本领域中已知的任何合适手段来进行多个受试者的基因组样本的数据集的获取和处理。
本公开还至少部分地基于以下认识:通过确定数据集中的患病者和未患病者来生成系谱以及精确所述系谱以形成富集系谱的信息对于下游分析发现罕见遗传变异与疾病之间的联系是尤其至关重要的。
可以通过以下方式来定义数据集中的患病者:基于至少一种二元性状或极端数量性状或它们的组合的存在来识别数据集中的个体。
在一些示例性实施方案中,使用来自国际疾病和相关健康问题统计分类列表(ICD)的三字母代码来定义二元性状。在一些特定的示例性实施方案中,用来自ICD的第9次或第10次修订版的三字母代码来定义二元性状。还可以使用来自ICD的第9次或第10次修订版的四字母代码来定义二元性状。如果个体的表型具有所描述的二元性状,则可以将该个体确定是“患病者”。在一些示例性实施方案中,即使先前被确定是“患病者”,在该同期群中具有二元性状并且流行率超过5%的个体也可以被确定是“未患病者”。此外,如果个体在病历中有对该性状是否存在的指示,并且如果该个体具有矛盾的记录,则该个体被确定是未知患病者。
在一些示例性实施方案中,通过以下方式来定义极端数量性状:基于群体中性状的分布来选取具有该性状的极高或极低值的个体,例如计算每个性状值的z得分,并且如果个体性状关于极高或极低性状值的z得分分别高于2或低于-2,则将该个体标记为“患病者”。此外,如果个体在病历中有对该性状是否存在的指示,并且如果该个体具有矛盾的记录,则该个体被确定是未知患病者。
包括患病者的系谱可进一步精确以生成富集系谱。可以基于表型隔离或p值来富集系谱。
图1是示例性实施方案的流程图,其中来自一级网络的个体被确定是患病者和未患病者。在步骤100处通过任何适当的手段从多个人类受试者生成个体的一级网络。可以在110处针对每种记录的二元性状或每种记录的数量性状或它们的组合来评估网络中的每个个体。可以在步骤120处针对每种记录的二元性状评估网络中的每个个体,并且如果个体受到所述二元性状的影响,则在步骤140将所述个体分类为“患病者”。相反,如果个体未受到所考虑的特定二元性状的影响,则在步骤150处将该个体分类为“未患病者”。可以在步骤130处针对每种记录的数量性状评估网络中的每个个体,并且如果个体受到所述数量性状的影响,则在步骤140将所述个体分类为“患病者”。相反,如果个体未受到所考虑的特定数量性状的影响,则在步骤150处将该个体分类为“未患病者”。
图2是另一示例性实施方案的流程图,其中来自一级网络的个体被确定是患病者和未患病者。在通过任何合适的手段在步骤100处从多个人类受试者生成个体的一级网络之后,可以在110处针对每种记录的二元性状或每种记录的数量性状或它们的组合来评估网络中的每个个体。此外,在步骤155处基于二元性状或数量性状的存在来评估具有任何记录的二元性状或每个记录的数量性状或它们的组合的每个个体。在步骤155之后,步骤160可以对个体进行分类:如果用于将个体分类为患病者的二元性状在同期群中的流行率超过5%,则可以在步骤190处将患病者分类为“未患病者”;并且如果用于将个体分类为患病者的二元性状的流行率低于5%,则可以在步骤180处将患病者分类为“患病者”。类似地,步骤170可以对个体进行重新分类:如果用于将个体分类为患病者的数量性状相比于同期群的平均数量性状大两个标准差,则在步骤180处将该个体分类为“患病者”,否则在步骤190处将该个体分类为“未患病者”。
系谱之内或系谱之间的表型隔离可以生成显性和加性隔离模型或隐性隔离模型。在将表型隔离成显性和加性隔离模型的系谱的一些示例性实施方案中,具有一种可能结构和多个三个有共同祖先的患病者的系谱可以用于生成富集系谱。此外,通过选择具有一个或多于一个相关未患病者的系谱以减少假阳性,可以对富集系谱进行优先级排序以供进行隔离分析。
在将表型隔离成隐性隔离模型的系谱的一些示例性实施方案中,将具有一种可能结构和多于一个具有未患病亲本的患病者的系谱用于生成富集系谱。此外,可以通过选择具有两个或多于两个患病同胞的系谱,来对富集系谱进行优先级排序以供进行隔离分析。
在一些示例性实施方案中,可以将来自两个或更多个表型相似或互补的二元或极端数量性状的患病者合并以形成涵盖所有这些性状的疾患的患病者。例如,当寻找针对双相障碍富集的系谱时,也可以考虑单相障碍,因为双相障碍的遗传原因可能在一些个体中仅表现为单相的。
在一些示例性实施方案中,可以选择具有两个或更多个极端或令人感兴趣的二元或极端数量性状的患病者,以形成涵盖所有这两种或更多种性状的疾患的患病者。取具有两种或更多种极端或令人感兴趣的性状的患病者的交集,可识别个体的更同质的子集。例如,为了获得具有患哮喘和COPD两者的个体的富集系谱,将患有哮喘和COPD两者的患者的交集视为患病者。
应当理解的是,本公开不限于上述疾患或隔离模型中的任一者,并且可以基于至少一种二元性状、极端数量性状或它们的组合针对任何疾患或隔离模型进行系谱富集。
或者,可以基于p值确定富集系谱。在一些示例性实施方案中,在识别系谱的首建者锚定分支时,进行二项式检验以评估系谱是否是针对二元性状富集的。在其他示例性实施方案中,在识别系谱的首建者锚定分支时,执行t检验以评估系谱是否是针对极端数量性状富集的。此外,设置了多重检验校正p值截止值以去除假阳性。
本公开至少部分地基于以下认识:对于针对具有给定表型的患病个体富集的系谱,伴随的(例如,罕见的)变异可以与感兴趣的表型隔离并驱动所述感兴趣的表型。由于此类遗传原因可能更可能在家族单元内共享,因此识别针对具有感兴趣的表型的患病者富集的系谱可以帮助识别驱动这些表型的偶然(例如,罕见)突变。
一旦已经识别出了富集系谱,就可以通过进行隔离分析和基于家族的关联分析来确定潜在的遗传原因。对于一些系谱,将存在与患病者隔离的已知致病突变。剩余系谱可以按照跨多个系谱在患病者中隔离的变异和基因进行优先级排序,或者使用数据集中不包含在系谱中的患病者进行优先级排序。无论如何,来自这些隔离分析的结果可包括候选变异的列表。
可以通过测试不同普遍度的模型来执行隔离分析。可以将具有各种限制(例如,显性或隐性遗传)的模型与最通用的模型进行比较,其中估计模型中的所有参数以查看哪种模型与数据最拟合。具有大系谱和许多患病个体的的家族对于建立所述基因和识别特定基因都是尤其信息丰富的。
使用系谱结构帮助识别给定表型的遗传原因的方法通常涉及关联定位、连锁分析或两者上的创新变异。此类方法包括MORGAN、pVAAST、FBAT(www.hsph.harvard.edu/fbat/fbat.htm)、QTDT(csg.sph.umich.edu/abecasis/qtdt/)、ROADTRIPS、rareIBD和RV-GDT。要使用的适当方法取决于表型、遗传模式、祖先背景、系谱结构/大小、系谱数目和无关数据集的大小。除了使用关系和系谱直接查询基因-表型关联外,所述关系和系谱还可以以多种其他方式用于生成附加或改进的数据:系谱感知填充、系谱感知定相、孟德尔错误检查、复合杂合敲除检测和从头突变调用(calling),以及变异调用验证。
本发明描述或例示的方法中的任何方法都可以作为计算机实现的方法和/或系统来实践。为此目的,可以使用本领域普通技术人员已知的任何合适的计算机系统。
图3示出了可以在其中操作本发明方法和系统的示例性环境200的各个方面。本发明方法可以在采用数字和模拟设备两者的各种类型的网络和系统中使用。本文提供了功能描述,并且相应功能可以由软件、硬件或软件和硬件的组合来执行。
环境200可以包括本地数据/处理中心210。本地数据/处理中心210可以包括一个或多个网络,例如局域网,以促进一个或多个计算设备之间的通信。一个或多个计算设备可以用于存储、处理、分析、输出和/或可视化生物数据。环境200可以可选地包括医学数据提供者220。医学数据提供者220可以包括一个或多个生物数据源。例如,医学数据提供者220可以包括一个或多个健康系统,该一个或多个健康系统有权访问一个或多个患者的医学信息。医学信息可以包括例如病史、医学专业观察和评论、实验室报告、诊断、医嘱、处方、生命体征、体液平衡、呼吸功能、血液参数、心电图、x射线、CT扫描、MRI数据、实验室测试结果、诊断、预后、评估、入院和出院记录、以及患者注册信息。医学数据提供者220可以包括一个或多个网络,例如局域网,以促进一个或多个计算设备之间的通信。一个或多个计算设备可以用于存储、处理、分析、输出和/或可视化医学信息。医学数据提供者220可以对医学信息进行去识别化,并且可以将所去识别化的医学信息提供给本地数据/处理中心210。去识别化的医学信息可以包括每个患者的唯一标识符,以便在将医学信息保持在去识别化状态的同时将一个患者与另一个患者的医学信息区分开。去识别化的医学信息阻止将患者的身份与其特定医学信息相关联。本地数据/处理中心210可以分析去识别化的医学信息,以为每个患者分配一个或多个表型(例如,通过分配国际疾病分类“ICD”和/或当前程序术语“CPT”代码)。
环境200可以包括NGS测序设施230。NGS测序设施230可包含一个或多个测序仪(例如,Illumina HiSeq 2500,Pacific Biosciences PacBio RS II等)。一个或多个测序仪可被配置用于外显子组测序、全外显子组测序、RNA-seq、全基因组测序、靶向测序等。在一个示例性方面中,医学数据提供者220可以提供来自与该去识别化的医学信息相关联的患者的生物样本。唯一标识符可用于维持生物样本和与该生物样本对应的去识别化的医学信息之间的关联。NGS测序设施230可以基于生物样本对每个患者的外显子组进行测序。为了在测序之前存储生物样本,NGS测序设施230可包含生物库(例如,来自LiconicInstruments)。可以在试管(每个与患者相关联的试管)中接纳生物样本,每个试管可包括条形码(或其他标识符),可以扫描所述条形码以自动将样本记录到本地数据/处理中心210中。NGS测序设施230可包括一个或多个机器人,该一个或多个机器人用于测序的一个或多个阶段中以确保统一的数据和有效的不间断操作。因此,NGS测序设备230可以每年对数万个外显子组进行测序。在一个方面中,NGS测序设施230具有每月对至少1000个、2000个、3000个、4000个、5000个、6000个、7000个、8000个、9000个、10,000个、11,000个或12,000个全外显子组进行测序的功能能力。
可以将NGS测序设施230生成的生物数据(例如,原始测序数据)传送到本地数据/处理中心210,该本地数据/处理中心可随后将该生物数据传送到远程数据/处理中心240。远程数据/处理中心240可以包括基于云的数据存储和处理中心,该基于云的数据存储和处理中心包括一个或多个计算设备。本地数据/处理中心210和NGS测序设施230可以直接通过一条或多条高容量光纤线路将数据传送至远程数据/处理中心240和从该远程数据/处理中心传送数据,但是也可以考虑其他数据传送系统(例如,互联网)。在示例性方面中,远程数据/处理中心240可包括第三方系统,例如亚马逊网络服务(DNAnexus)。远程数据/处理中心240可以促进分析步骤的自动化,并允许以安全的方式与一个或多个合作者250共享数据。在从本地数据/处理中心210接收到生物数据时,远程数据/处理中心240可以使用生物信息学工具执行主要和次要数据分析的一系列自动化管线步骤,从而产生每个样本的带注释的变异文件。可以将来自此类数据分析的结果(例如,基因型)传递回本地数据/处理中心210,并且例如集成到实验室信息管理系统(LIMS)中,该LIMS可被配置用于维持每个生物样本的状态。
然后,本地数据/处理中心210可以利用经由NGS测序设施230和远程数据/处理中心240获得的生物学数据(例如,基因型)结合去识别化的医学信息(包括所识别的表型)来识别基因型与表型之间的关联。例如,本地数据/处理中心210可以应用表型优先方法,其中定义了在某些疾病区域中可能具有治疗潜力的表型,例如心血管疾病的血脂极端。另一个示例是研究肥胖患者,以识别似乎可以受保护免受典型范围的合并症影响的个体。另一种方法是从基因型和假设开始,例如基因X参与导致疾病Y或保护免受疾病Y。
在示例性方面中,一个或多个合作者250可以经由网络诸如因特网260访问生物数据和/或去识别化的医学信息中的一些或全部。
在示例性方面中,在图4中示出,本地数据/处理中心210和/或远程数据/处理中心240中的一者或多者可以包括一个或多个计算设备,该一个或多个计算设备包括遗传数据部件300、表型数据部件310、遗传变异-表型关联数据部件320、和/或数据分析部件330中的一者或多者。遗传数据部件300、表型数据部件310和/或遗传变异-表型关联数据部件320可被配置用于以下中的一者或多者:序列数据的质量评定、与参考基因组的读段比对、变异识别、变异注释、表型识别、变异-表型关联识别、数据可视化、它们的组合等。
在示例性方面中,部件中的一个或多个部件可以采取完全硬件实施方案,完全软件实施方案,或组合软件和硬件方面的实施方案的形式。此外,这些方法和系统可以采取计算机可读存储介质上的计算机程序产品的形式,该计算机可读存储介质具有体现在该存储介质中的计算机可读程序指令(例如,非暂时性计算机软件)。更具体地,本发明的方法和系统可以采取网络实现的计算机软件的形式。可以利用任何合适的计算机可读存储介质,包括硬盘、CD-ROM、光学存储设备或磁性存储设备。
在示例性方面中,遗传数据部件300可被配置用于对一个或多个遗传变异进行功能注释。遗传数据部件300还可被配置用于对一种或多种遗传变异进行存储、分析、接收等。可以从自一个或多个患者(受试者)获得的序列数据(例如,原始序列数据)注释所述一个或多个遗传变异。例如,可以从至少100,000个、200,000个、300,000个、400,000个或500,000个受试者中的每一个受试者注释所述一个或多个遗传变异。对一个或多个遗传变异进行功能注释的结果是遗传变异数据的生成。举例来说,遗传变异数据可包括一个或多个变异调用格式(VCF)文件。VCF文件是用于表示SNP、插入缺失和/或结构变异调用的文本文件格式。评定变异对转录本/基因的功能影响,并识别潜在的功能丧失(pLoF)候选者。使用Ensembl75基因定义用snpEff对变异进行注释,然后针对每个变异(和基因)对所述功能注释进行进一步处理。
如本文所提供的用数字和/或字母对方法步骤进行连续标记并不意味着将方法或其任何实施方案限制为特定的所指示次序。
在整个说明书中引用了各种出版物,包括专利、专利申请、公开的专利申请、登录号、技术文章和学术文章。这些引用的参考文献中的每一篇参考文献出于所有目的以引用方式整体并入本文。
将通过参考以下实施例更充分地理解本公开,提供以下实施例以更详细地描述本公开。它们旨在说明而不应解释为限制本公开的范围。
实施例
对93,368位去识别化的Geisinger健康系统(Geisinger Health System,GHS)参与者(已同意加入MyCode社区健康计划(MyCode Community Health Initiative))进行测序。作为这项计划的一部分,个体同意提供血液和DNA样本用于广泛的未来研究,包括进行作为Regeneron GHS DiscovEHR合作的一部分的基因组分析,并根据Geisinger机构审查委员会(Geisinger Institutional Review Board)批准的协议与GHS EHR中的数据链接。根据参与者的同意和IRB的批准进行所有执行的分析。每个参与者的外显子组都与对应的去识别化的EHR链接。DiscovEHR研究并未专门针对家族作为研究参与者,而是隐含地富集了因为慢性健康问题而频繁与医疗卫生系统互动的成人(和可能彼此相关的成人)以及冠状动脉导管插入实验室(Coronary Catheterization Laboratory)和来自GHS的减肥服务的参与者。
先前已经描述了前61K样本(“VCRome集合”)的样本制备和测序(Dewey等人,Science(2016)354:aaf6814)。以相同的方法制备具有31K样本的剩余集合,不同之处在于代替NimbleGen探测捕获,使用IDT的xGen探针的稍加修改版本,并添加补充探针来捕获基因组的被NimbleGen VCRome捕获试剂很好覆盖但被标准xGen探针不良覆盖的区域。将捕获的片段与链霉亲和素缀合的珠粒结合,并根据制造商(IDT)推荐的方案通过一系列严格的洗涤去除非特异性DNA片段。第二组样本称为“xGen集合”。使用基因组分析工具包(GATK;Web Resources)产生变异调用。GATK用于进行对每个样本的在推定插入缺失周围的经比对的、重复标记的读段的局部重新比对。使用GATK的HaplotypeCaller处理经插入缺失重新比对的、重复标记的读段,以识别样本与基因组变异调用格式(gVCf)的基因组参考不同的所有外显子位置。使用GATK的GenotypeGYCF完成每个样品的基因分型,并且具有50个随机选择的样本的训练集输出单样本变异调用格式(VCF)文件,从而识别与参考相比的单核苷酸变异(SNV)和插入缺失。使用单样本VCF文件创建伪样本,该伪样本包含来自两个集合中的单样本VCF文件的所有可变位点。通过使用伪样本联合调用200个单样本gVCF文件来为VCRome集创建独立pVCF文件,以迫使每个样本在跨两个捕获集合的所有可变位点处有调用或无调用。合并所有200个样本的pVCF文件以创建VCRome pVCF文件,然后重复此过程以创建xGen pVCF文件。将VCRome和xGen pVCF文件组合在一起以创建联合pVCF。将序列读段与GRCh38进行比对,并通过使用Ensembl 85基因定义进行变异注释。基因定义仅限于54,214个转录本,对应于19,467个基因,这些基因是编码蛋白质的,具有注释的起始和终止处。经过前述的样品质控处理后,仍有92,455个外显子组需要分析。
使用PLINKv1.910将联合数据集与HapMap318合并,并根据参考SNP簇(duster)ID保留两个数据集中的SNP。通过应用以下PLlNK过滤器,该分析仅限于次要等位基因频率>10%、基因型缺失<5%并且Hardy-Weinberg平衡p值>0.00001的高质量共同SNP:“-maf0.1-geno 0.05-snps-only-h we 0.00001”。计算用于HapMap3样本的主分量(PC),然后通过使用PLINK将数据集中的每个样本投射到这些PC上。我们将PC用于HapMap3样本,以针对以下五个祖先超类中的每一个祖先超类训练内核密度估计器(KDE):非洲(AFR)、混合的美洲(AMR)、东亚(EAS)、欧洲(EUR)和南亚(SAS)。计算KDE以估计每个样本属于每个超类的可能性。对于每个样本,基于可能性分配祖先超类。如果一个样本有两个祖先组的可能性大于0.3,则将该样本分配为AFR超过EUR、AMR超过EUR、AMR超过EAS、SAS超过EUR以及AMR超过AFR;否则为“未知”。如果零个或多于两个祖先组具有足够高的可能性,则为样本分配“未知”祖先。具有未知祖先的样本被从基于祖先的按血统身份(IBD)计算排除。
通过使用以下标记在完整数据集上运行PLINK来过滤出高质量的常见变异:--maf0.1--geno 0.05--snps-only--hwe 0.00001。然后采用两管齐下的方法从外显子组数据获得准确的IBD估计。首先,在如根据祖先分析确定的同一祖先超类(例如AMR、AFR、EAS、EUR和SAS)内计算个体之间的IBD估计值。
其次,为了捕捉具有不同祖先的个体之间的一级关系,使用--min0.3PLINK选项在所有个体之间计算IBD估计值。然后将个体分组为一级家族网络,其中网络节点为个体并且边缘为一级关系。每个一级家族网络都通过prePRIMUS管线运行(Staples等人(2014);Am.J.Hum.Genet.95,553–564),这使样品的祖先与合适的祖先次要等位基因频率相匹配以改善IBD估计。此过程准确地估计了每个家族网络中的个体之间的一级关系(最小PI_HAT为0.15)。
从92,455名个体的DiscovEHR数据集,识别出了了43个单卵双胞胎、16,476个亲本-子代关系、10,479个全同胞关系和39,000个二级关系(图5)。将个体被视为节点并且将关系视为边,以生成无向图。仅使用一级关系,就识别出了12,594个连接的分量,这些连接的分量被称为一级家族网络。DiscovEHR同期群中的个体中的39%在数据集中具有至少一个一级亲属。
表1(DiscovEHR数据集的祖先分类)
表2(DiscovEHR数据集中参与一级关系的个体的祖先背景的完整分类)
用PRIMUSv1.9.0重建在DiscovEHR同期群中识别出的所有一级家族网络。将组合的IBD估计值与在遗传来源的性别和EHR报告的年龄一起提供给PRIMUS。指定PI_HAT>0.375的相关性截止,以将重建限制于一级家族网络。
从数据集中发现了300多种电子健康记录(EHR)来源的以孟德尔方式隔离的表型,从而提供了2,000多种潜在信息丰富的系谱-表型配对,该系谱-表型配对使得能够进行大规模传统孟德尔分析。
针对至少一种二元性状、极端数量性状或它们的组合将来自一级家族网络的个体确定是“患病者”或“未患病者”。将这些患病者集合与系谱取交集以识别出用足够多的患病个体富集的系谱,以便适合用于基于家族的隔离分析。
从数据集中识别出了2,978个性状-系谱富集对(2,596个显性和382个隐性)。在这些性状-系谱富集对中,在981个系谱中有3,975个具有1,015种不同性状的患病个体。性状中的超过50%富集于两个或更多个系谱中,并且357种性状富集于三个或更多个系谱中。
此外,在2,978个性状-系谱富集对中,1,911个是二元性状-系谱富集对,具有809个不同性状和673个系谱。在二元性状-系谱富集对中,最富集的系谱是针对龋齿的(N=46)。进一步在2,978个性状-系谱富集对中,1,067个是数量性状-系谱富集对,具有206个不同性状和581个系谱。在数量性状-系谱富集对中,最富集的系谱是针对高甘油三酸酯_Med_LabValue的(N=19)。
7.1原发性血栓形成
原发性血栓形成是遗传止血机制疾患,该疾患导致血栓形成(高凝状态)。这通常会影响静脉系统(例如,深静脉血栓形成、肺栓塞)。
基于针对原发性血栓形成的二元性状(Phe10_D685,ICD10 4D)将群体中的个体确定是患病者。
使用实施例6中所述的方法从重建的系谱(表3和表4)中过滤出一级系谱,以去除所有不是只有一种可能结构并且具有少于三个具有共同祖先的原发性血栓形成患病者的系谱,以产生原发性血栓形成的富集系谱。在该同期群中,原发性血栓形成(Phe10_D685,ICD10CM D68.5)的流行率为1.3%。
因此识别出了几个针对原发性血栓形成富集的系谱(参见图6)。
表3
表4
7.2遗传性出血性毛细血管扩张
遗传性出血性毛细血管扩张(HTT)是一种罕见的常染色体显性疾患,其影响全身血管(导致血管发育不良)并导致出血倾向。(该病症也称为Osler-Weber-Rendu病(OWRD);这两个术语可以互换使用。)HHT由粘膜皮肤毛细血管扩张(mucocutaneoustelangiectase)和动静脉畸形(AVM)表现出来,是严重发病和死亡的潜在原因。病变可影响鼻咽、中枢神经系统(CNS)、肺、肝和脾脏,以及泌尿道、胃肠(GI)道、结膜、躯干、手臂和手指。
基于针对HTT(Phe10_I780,ICD10CM I78.0)的二元性状,确定群体中的个体为患病者。
使用实施例6中所述的方法针对HTT重建两个系谱(参见表5和表6)。两个系谱都具有三个具有共同祖先的HHT患病者和一种可能结构。此外,在该同期群中,HTT的流行率为0.0%。
表5
表6
使用针对HTT的二元性状富集的两个系谱来执行罕见变异隔离分析(参见图7A和图7B)。
对于图7A中显示的针对HTT富集的系谱,隔离和关联分析表明,SMAD4基因变异与系谱(参见表7)中的HTT表型共隔离。SMAD4(SMAD家族成员4)是SMAD信号转导蛋白家族的成员。Smad蛋白响应于转化生长因子(TGF)-β信号传导而由跨膜丝氨酸-苏氨酸受体激酶磷酸化并激活。SMAD4与其他活化的Smad蛋白形成同源复合物和异源复合物,所述复合物随后在细胞核中积累并调节靶基因的转录,并且SMAD4是BMP信号传导通路的重要部件。SMAD4的突变或缺失已与遗传性疾患遗传性出血性毛细血管扩张综合征(HHT)和Myhre综合征;以及家族性癌症易感性疾患,包括青少年息肉病综合征(染色体18q21上的SMAD4基因杂合突变)相关联。SMAD4充当肿瘤抑制因子并抑制上皮细胞增殖。它还可通过减少血管生成和增加血管超通透性而对肿瘤具有抑制效应。在胰腺癌中已经识别出了SMAD4中的体细胞突变。
表7
对于图7B和图8中显示的针对HTT富集的系谱,隔离和关联分析表明,激活素A受体II型样1(ACVRL1)基因的变异与所述系谱中的HTT表型共隔离(参见表8)。ACVRL1基因编码TGF-β配体超家族的I型细胞表面受体,并与形成受体丝氨酸/苏氨酸激酶亚家族的其他紧密相关的ALK或激活素受体样激酶蛋白共享相似的结构域结构。ACVRL1的突变与2型出血性毛细血管扩张相关联,也称为Rendu-Osler-Weber综合征2和肺动脉高压。患者患有结膜毛细血管扩张、通常导致作为疾病首发病征鼻出血的鼻粘膜毛细血管扩张、口腔毛细血管扩张、各种器官中的动静脉畸形、皮肤毛细血管扩张,贫血,并且一些患者发展为肺动脉高压。HHT2的内脏发现包括肺动静脉畸形(PAVM)、脑AVM、脊柱AVM、肝AVM、归因于AVM的胃肠道出血,以及肝硬化。HHT2的神经系统表现包括癫痫发作、缺血性中风、偏头痛、脑动静脉畸形和脑出血。
表8.
7.3通过肺活量测量法测量的慢性阻塞性肺病(GOLD)2-4级患者的肺气肿
肺气肿是一种导致呼吸短促的肺病症,并且是包括慢性阻塞性肺疾病(COPD)在内的疾病之一。在患有肺气肿的人中,肺中的呼吸囊(肺泡)受损。随着时间的推移,呼吸囊的内壁会减弱并破裂,从而形成较大的空气空间,而不是许多较小的空气空间。这减小了肺部的表面积,并进而减少可到达血液中的氧气量。在呼气时,受损的肺泡无法正常工作,并且旧空气被困住,没有留下空间让新鲜的富氧空气进入。
从肺功能检查的数量性状得出“通过肺活量测量法测量的GOLD2-4级患者的肺气肿”的二元性状。使用基于在非吸烟COPD患者的电子病历中报告的多发性发病率的所述患者的高置信度集合。肺功能测试的数量性状之一是使用“根据最近的肺活量测量法将支气管扩张剂前在50%的强制肺活量下的强制呼气流量转换为在50%的强制肺活量下的强制吸气流量”定义的。群体中该性状的平均值为0,并且标准偏差为0.27。使用数量性状的下限执行富集。肺功能测试的另一种数量性状是使用“在距最近的肺活量测量法1秒内预测的支气管扩张剂后强制呼气量的百分比”定义的。群体中该性状的平均值为81.89,并且标准偏差为20.84。使用数量性状的下限执行富集。
从一级家族网络中分离出针对通过肺活量测量法测量的GOLD2-4级患者的肺气肿二元性状富集的系谱(参见图9)。在该同期群中,该特定表型的流行率为1.8%。所述系谱仅具有一个可能结构,并且包含三个具有共同祖先的患病者。
7.4肾脏移植
从一级家族网络中分离出针对肾脏移植二元性状(Phe9_V420,ICD9DM V42.0)富集的系谱。该特定表型的流行率为0.8%。
一级系谱仅具有一种可能结构,并且有四个具有共同祖先的患病者。识别出包含所需标准的系谱(参见图10和表9)。
表9
7.5终末期肾脏疾病
根据终末期肾脏疾病的二元性状(Phe10_5856,ICD9CM 585.6)将群体中的个体确定是患病者。识别出几个针对终末期肾脏疾病富集的系谱(图11)。
7.6遗传性运动和感觉神经病(Charcot-Marie-Tooth病)
Charcot-Marie-Tooth病(CMT)是最常见的遗传性神经系统疾患之一,在美国2,500人中有约1人患病。它也被称为遗传性运动和感觉神经病(HMSN)或腓骨肌萎缩症,包括一组影响周围神经的疾患。
基于遗传性运动和感觉神经病的二元性状(Phe10_G600,ICD10CM G60.0)将群体中的个体确定是患病者。在该同期群中,该特定表型的流行率为0.1%。
根据从实施例6重建的系谱,针对遗传性运动和感觉神经病的一级系谱具有一个可能结构和三个具有共同祖先的患病者(参见图12和表10)。
表10.
对于针对遗传性运动和感觉神经病富集的系谱,隔离和关联分析表明,原肌球蛋白2(β)(TPM2)基因变异与系谱中的遗传性运动和感觉神经病表型共隔离(表11)。TPM2编码β-原肌球蛋白,β-原肌球蛋白为肌动蛋白丝结合蛋白家族的成员,并且主要在慢速1型肌纤维中表达。TPM2中的突变可改变其他肌节原肌球蛋白的表达,并引起帽病(cap disease)、线状体肌病和远端关节弯曲综合征。
表11.
在各种组织中编码的TPM2的每百万转录本(TPM)的基因表达数据表明了在动脉、乙状结肠、食道-胃肠道连接处、食道-肌层和骨骼肌中的高发生率(参见图13)。
系谱中的患病者的患者记录(参见表12),表明这个家族并没有显示出遗传性运动和感觉神经病的迹象,而是他们患有由于TPM2突变导致的线状体肌病4型(Donner等人,Neuromuscular Disorders(2009)19:348-3351)。
表12.
7.7双相障碍
双相障碍或“躁郁症”会导致极端的情绪变化,包括情绪高涨(躁狂或轻躁狂)和情绪低落(抑郁)。在任何给定年中,群体中的约2.6%(570万美国成人)患有这种疾患。
根据双相障碍和单相障碍将群体中的个体确定是患病者。双相障碍的ICD 10代码为F31;ICD 9代码为296.4至296.7。患者的子集(35%至40%)接受了锂处方。单相/重度抑郁疾患的ICD 10代码为F32、F33、F39;ICD-9代码为296.2/.3/.9(家族网络内继发的)。患有自闭症(ICD-10代码F84)和智力迟钝(ICD-10代码F70.9、F71.9、F72.9、F73.9、F79.9)的患者被排除出患病者组。该同期群中针对双相障碍(F319-3.2%)和单相障碍(F31、F32和F33-0.0%、4.1%和2.1%)的二元性状的流行率均低于5%。
从一级家族网络中分离出针对双相障碍的二元性状富集的系谱。
对一级系谱进行评估,以确保它仅具有一种可能结构,并且具有至少3个具有共同祖先的患病者(参见图14)。对富集系谱执行的隔离分析产生了与表型共隔离的可能变异的列表(表13)。与表型共隔离的变异C20orf203是有害的并且是非保守的。
表13.
已将FLJ33706(替代基因符号C20orf203)识别为引起尼古丁成瘾的可能原因。在各种组织中编码,但主要在小脑半球和脑小脑中表达的20号染色体开放阅读框203(C20orf203)的每百万转录本(TPM)的基因表达数据(图15)。连锁研究已识别出rs17123507(一种位于FLJ33706的3'UTR中的SNP)与尼古丁成瘾易感性显著相关联(Li等人,PLoSComputational Biology(2010)6:e1000734)。
此外,识别出了两个更富集的系谱(参见图16和图17;表14和表15)。这两个系谱都仅具有一个可能结构,并且具有超过三个具有共同祖先的患病者。
表14
表15
另外,另一针对双相障碍的二元性状富集的系谱仅具有一种可能结构,并且具有多于三个具有共同祖先的患病者(参见图18)。
对富集系谱执行的变异分析产生了与表型共隔离的可能变异的列表(表16)。
表16.
在表17列出的变异中,小头蛋白1(MCPH1)是报告的先天性小头畸形致病变异。在各种组织中编码的MCPH1的每百万转录本(TPM)的基因表达数据表明了在几种组织中的高发生率(参见图19)
1型先天性小头畸形的特征是头围比年龄相关平均值低超过3个标准偏差。脑重量显著减少,并且大脑皮层不成比例地小。患病的个体具有严重的智力障碍。一些MCHP1患者还表现出生长迟缓、身材矮小和染色体凝缩失调,如由在细胞遗传学制备物中检测到的大量前期样细胞和质量较差的中期G显带所表明的。
表17.
7.8地中海贫血
地中海贫血是一种遗传性血液疾患,其特征为体内的血红蛋白和红血细胞少于正常。地中海贫血的低血红蛋白和较少血红细胞可能引起贫血,从而使患者感到疲劳。
地中海贫血的ICD 10代码为D56。
从一级家族网络中分离出针对地中海贫血的二元性状富集的系谱。
对一级系谱进行评估,以确保它仅具有一种可能结构,并且具有至少3个具有共同祖先的患病者(参见图20)。识别出两个富集系谱(参见图20)。这两个系谱都仅具有一种可能结构,并且具有三个或更多个患病者。
对富集系谱执行的变异分析产生了与表型共隔离的可能HBB基因变异的列表。HBB基因提供了关于制造被称为β-珠蛋白的蛋白质的使用说明。β-珠蛋白是被称为血红蛋白的较大蛋白质的组分(亚基),它位于红血细胞内。在成人中,血红蛋白通常由四个蛋白亚基组成:β-珠蛋白的两个亚基和另一种称为α-珠蛋白的蛋白的两个亚基,α-珠蛋白的两个亚基是由另一种称为HBA的基因产生的。这些蛋白质亚基中的每一者都附接(结合)至称为血红素的含铁分子;每个血红素都在其中心处含有可与一个氧分子结合的铁分子。红学细胞内的血红蛋白与肺中的氧分子结合。这些细胞然后穿过血流,并将氧气递送到全身的组织。与HBB基因相关联的疾病包括β-地中海贫血和镰状细胞性贫血。
在HBB基因中识别出的与表型共隔离的两个突变是Gln40处的终止增益突变和Gly84处的移码突变(关联分析p值<3.1×10
7.10降低的碱性磷酸酶门诊患者中心趋势值
在医院中,既出于诊断有症状患者的目的,也出于筛查无症状患者的目的,非常频繁地对碱性磷酸酶进行常规实验室测试。尽管碱性磷酸酶存在于整个人体的各组织中,但是其在患有肝病和骨病的患者中最常见。
创建针对降低的碱性磷酸酶水平富集的系谱,并对其进行评估,以确保其仅具有一种可能结构并且具有至少三个具有共同祖先的患病者(参见图21)。
对富集系谱执行的变异分析表明,ALPL基因的错义突变与表型共隔离。ALPL基因提供了制备被称为组织非特异性碱性磷酸酶(TNSALP)的酶的使用说明。该酶在骨骼和牙齿的生长和发育中起重要作用。它还在许多其他组织中有活性,特别是在肝脏和肾脏中。该酶起磷酸酶的作用,这意味着它可以从其他分子中去除氧和磷原子(磷酸根基团)的簇。TNSALP对于矿化过程至关重要,在矿化过程中,矿物质诸如钙和磷沉积在发育中的骨骼和牙齿中。矿化对于强健而坚硬的骨头以及可承受咀嚼和磨碎的牙齿的形成至关重要。ALPL基因中识别出的杂合错义突变位于Leu275(Leu275Pro)处(参见图21)(关联分析p值为<7.2×10
- 用于在系谱内进行系谱富集和基于家族的分析的方法和系统
- 用于系谱分析的系统、方法和计算机程序产品