一种环戊烷并香豆素类化合物及其制备方法
文献发布时间:2023-06-19 11:42:32
技术领域
本发明涉及有机化合物合成领域,特别涉及一种环戊烷并香豆素类化合物及其制备方法。
背景技术
香豆素类以及环戊烷类化合物广泛存在于天然产物当中,并表现出一系列生物与医药活性(Med.Res.Rev.2003,23,322;Nat.Prod.Rep.2015,32,1472;Drugs,2002,62,107),而将两类骨架联合形成的环戊烷并香豆素类衍生物也表现出一定的活性,例如广为人知的黄曲霉素就为此类化合物的衍生物(Pharmac.Ther.1995,65,163.),此外Herbertenolide属于剪叶苔烷型倍半萜。该家族倍半萜对植物病原性真菌的表现出显著的生长抑制活性,并有望开发为抗真菌农药(J.Chem.Soc.Perkin Trans.1,1986,701)。尽管环烷烃并香豆素类化合物的合成方法已有诸多报道(Tetrahedron,2001,57,9299;Org.Lett.2007,9,3069;Angew.Chem.Int.Ed.2018,57,17100;Chem.Commun.,2018,54,12702;Adv.Synth.Catal.,2020,362,1679。
但目前以简单原料温和高效地合成环戊烷并香豆素类化合物报道仍然较少。鉴于此,发展简易经济的合成方法以合成多样的环戊烷并香豆素类化合物是非常重要的。
发明内容
为了弥补现有技术的不足,本发明提供了一种环戊烷并香豆素类化合物及其制备方法。
本发明的技术方案为:
一种环戊烷并香豆素类化合物,其化学结构式如式(Ⅰ)所示:
其中,R
作为优选方案,所述环戊烷并香豆素类化合物由式(Ⅱ)和式(Ⅲ)所示的原料合成;
作为优选方案,R
所述环戊烷并香豆素类化合物的制备方法,在氮气保护条件下,环状氮杂双烯与3-乙酰基香豆素衍生物溶于有机溶剂,加入有机碱催化剂,进行环加成反应,得到环戊烷并香豆素类化合物。
进一步地,环状氮杂双烯的结构如式(Ⅱ)所示;3-乙酰基香豆素衍生物的结构如式(Ⅲ)所示;
作为优选方案,所述有机碱催化剂为1,8-二氮杂二环十一碳-7-烯、4-二甲氨基吡啶、三乙烯二胺、三乙胺中的一种或多种。
进一步地,所述有机碱催化剂为1,8-二氮杂二环十一碳-7-烯。以1,8-二氮杂二环十一碳-7-烯为催化剂时,对反应的催化活性最优。
作为优选方案,所述有机溶剂为甲醇、乙醚、四氢呋喃、二氯甲烷、二氯乙烷、苯、甲苯、氯仿、乙酸乙酯、DMSO和三氟甲苯中的一种或多种。
进一步地,所述有机溶剂为四氢呋喃。四氢呋喃作为溶剂时,收率最高,且后处理方便。
作为优选方案,环状氮杂双烯与3-乙酰基香豆素衍生物的投料摩尔比为1:1-3;所述有机碱催化剂的摩尔量为环状氮杂双烯摩尔量的1%-20%。
作为优选方案,环加成反应时间为12-48小时;反应温度为25℃-60℃。
本发明的有益效果为:
1、本发明采用有机碱催化[3+2]环加成方式进行反应,属于原子经济性反应,不涉及过渡金属催化,产品不会存在重金属残留;
2、本发明反应条件温和(室温下反应),反应速度快,操作简便,副反应少,便于分离提纯,且可放大量制备;
3、本发明为合成新的具有潜在生物活性的环戊烷并香豆素类化合物提供了一种新的方式,丰富了化合物的多样性。
4、本发明合成的化合物具有抗植物病原菌活性的特性。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其他的附图。
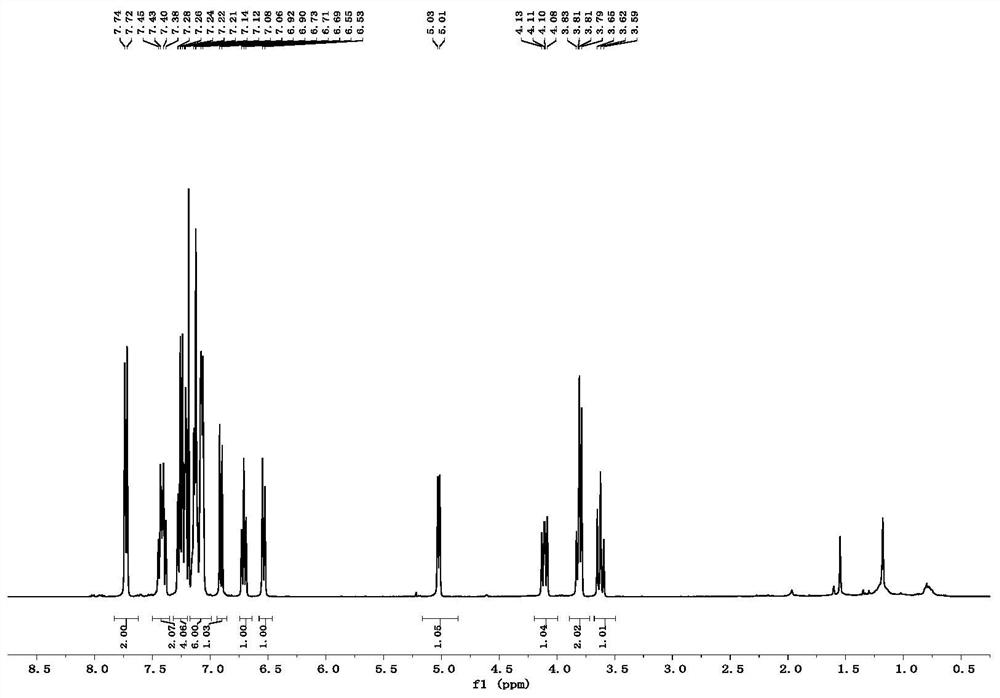
图1为实施例1所得环戊烷并香豆素类化合物3aa的核磁氢谱;
图2为实施例1所得环戊烷并香豆素类化合物3aa的核磁碳谱;
图3为实施例1所得环戊烷并香豆素类化合物3aa的高分辨质谱;
图4为实施例5所得环戊烷并香豆素类化合物3ba的核磁氢谱;
图5为实施例5所得环戊烷并香豆素类化合物3ba的核磁碳谱;
图6为实施例5所得环戊烷并香豆素类化合物3ba的高分辨质谱;
图7为实施例6所得环戊烷并香豆素类化合物3ca的核磁氢谱;
图8为实施例6所得环戊烷并香豆素类化合物3ca的核磁碳谱;
图9为实施例6所得环戊烷并香豆素类化合物3ca的高分辨质谱;
图10为实施例7所得环戊烷并香豆素类化合物3da的核磁氢谱;
图11为实施例7所得环戊烷并香豆素类化合物3da的核磁碳谱;
图12为实施例7所得环戊烷并香豆素类化合物3da的高分辨质谱;
图13为实施例8所得环戊烷并香豆素类化合物3ea的核磁氢谱;
图14为实施例8所得环戊烷并香豆素类化合物3ea的核磁碳谱;
图15为实施例8所得环戊烷并香豆素类化合物3ea的高分辨质谱;
图16为实施例8所得环戊烷并香豆素类化合物3fa的核磁氢谱;
图17为实施例8所得环戊烷并香豆素类化合物3fa的核磁碳谱;
图18为实施例8所得环戊烷并香豆素类化合物3fa的高分辨质谱;
注:实施例制备的各化合物均经过核磁氢谱、核磁碳谱以及高分辨率质谱的检测,由于谱图过多,不一一公布。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
根据前期试验,有机溶剂最优选为四氢呋喃,四氢呋喃作为溶剂时,收率最高,且后处理方便。采用其它有机溶剂作为溶剂时,收率都会有一定的降低(有机反应的溶剂效应),而且沸点高的有机溶剂浓缩处理时间较长,且浪费能源。因此实施例中均以四氢呋喃作为溶剂,但并不限定溶剂只能为四氢呋喃。
实施例1
将28.5mg(0.1mmol)化合物1a,41.2mg(0.12mmol)化合物2a和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入3.0mg(0.02mmol)1,8-二氮杂二环十一碳-7-烯混匀进行环加成反应。该反应体系中,化合物1a与化合物2a的摩尔比为1:1.2,1,8-二氮杂二环十一碳-7-烯占化合物1a的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到54.0mg产品3aa,收率85%。
实施例2
将28.5mg(0.1mmol)化合物1a,41.2mg(0.12mmol)化合物2a和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入2.2mg(0.02mmol)三乙烯二胺(DABCO)混匀进行环加成反应。该反应体系中,化合物1a与化合物2a的摩尔比为1:1.2,三乙烯二胺(DABCO)占化合物1a的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到35.8mg产品3aa,收率57%。
与实施例1相比,催化剂替换为DABCO,其它同实施例1。
实施例3
将28.5mg(0.1mmol)化合物1a,41.2mg(0.12mmol)化合物2a和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入2.4mg(0.02mmol)4-二甲氨基吡啶(DMAP)混匀进行环加成反应。该反应体系中,化合物1a与化合物2a的摩尔比为1:1.2,4-二甲氨基吡啶(DMAP)占化合物1a的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到39.0mg产品3aa,收率62%。
与实施例1相比,催化剂替换为DMAP,其它同实施例1。
实施例4
将28.5mg(0.1mmol)化合物1a,41.2mg(0.12mmol)化合物2a和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入2.0mg(0.02mmol)三乙胺(Et
与实施例1相比,催化剂替换为三乙胺,其它同实施例1。
实施例1-实施例4所得产品结构相同,产品3aa的核磁氢谱、核磁碳谱和高分辨质谱结果如下:
HRMS(ESI)calcd for C
由实施例1-4可知,几种有机碱中,1,8-二氮杂二环十一碳-7-烯催化活性最高,且以1,8-二氮杂二环十一碳-7-烯为催化剂时,产率远远优于其它三种有机碱催化剂。
实施例5
将30.3mg(0.1mmol)化合物1b,41.2mg(0.12mmol)化合物2a和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入3.0mg(0.02mmol)1,8-二氮杂二环十一碳-7-烯混匀进行环加成反应。该反应体系中,化合物1b与化合物2a的摩尔比为1:1.2,1,8-二氮杂二环十一碳-7-烯占化合物1b的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到55.0mg产品3ba,收率85%。
产品3ba的核磁氢谱、核磁碳谱和高分辨质谱结果如下:
HRMS(ESI)calcd for C
实施例6
将30.3mg(0.1mmol)化合物1c,41.2mg(0.12mmol)化合物2a和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入3.0mg(0.02mmol)1,8-二氮杂二环十一碳-7-烯混匀进行环加成反应。该反应体系中,化合物1c与化合物2a的摩尔比为1:1.2,1,8-二氮杂二环十一碳-7-烯占化合物1c的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到58.2mg产品3ca,收率90%。
产品3ca的核磁氢谱、核磁碳谱和高分辨质谱结果如下:
HRMS(ESI)calcd for C
实施例7
将30.3mg(0.1mmol)化合物1d,41.2mg(0.12mmol)化合物2a和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入3.0mg(0.02mmol)1,8-二氮杂二环十一碳-7-烯混匀进行环加成反应。该反应体系中,化合物1d与化合物2a的摩尔比为1:1.2,1,8-二氮杂二环十一碳-7-烯占化合物1d的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到55.0mg产品3da,收率85%。
产品3da的核磁氢谱、核磁碳谱和高分辨质谱结果如下:
HRMS(ESI)calcd for C
实施例8
将32.0mg(0.1mmol)化合物1e,41.2mg(0.12mmol)化合物2a和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入3.0mg(0.02mmol)1,8-二氮杂二环十一碳-7-烯混匀进行环加成反应。该反应体系中,化合物1e与化合物2a的摩尔比为1:1.2,1,8-二氮杂二环十一碳-7-烯占化合物1e的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到57.0mg产品3ea,收率86%。
产品3ea的核磁氢谱、核磁碳谱和高分辨质谱结果如下:
HRMS(ESI)calcd for C
实施例9
将32.0mg(0.1mmol)化合物1f,41.2mg(0.12mmol)化合物2a和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入3.0mg(0.02mmol)1,8-二氮杂二环十一碳-7-烯混匀进行环加成反应。该反应体系中,化合物1f与化合物2a的摩尔比为1:1.2,1,8-二氮杂二环十一碳-7-烯占化合物1f的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到53.0mg产品3fa,收率80%。
产品3fa的核磁氢谱、核磁碳谱和高分辨质谱结果如下:
HRMS(ESI)calcd for C
实施例10
将36.4mg(0.1mmol)化合物1g,41.2mg(0.12mmol)化合物2a和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入3.0mg(0.02mmol)1,8-二氮杂二环十一碳-7-烯混匀进行环加成反应。该反应体系中,化合物1g与化合物2a的摩尔比为1:1.2,1,8-二氮杂二环十一碳-7-烯占化合物1g的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到56.6mg产品3ga,收率80%。
产品3ga的核磁氢谱、核磁碳谱和高分辨质谱结果如下:
HRMS(ESI)calcd for C
实施例11
将29.9mg(0.1mmol)化合物1h,41.2mg(0.12mmol)化合物2a和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入3.0mg(0.02mmol)1,8-二氮杂二环十一碳-7-烯混匀进行环加成反应。该反应体系中,化合物1h与化合物2a的摩尔比为1:1.2,1,8-二氮杂二环十一碳-7-烯占化合物1h的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到50.8mg产品3ha,收率79%。
产品3ha的核磁氢谱、核磁碳谱和高分辨质谱结果如下:
HRMS(ESI)calcd for C
实施例12
将29.9mg(0.1mmol)化合物1i,41.2mg(0.12mmol)化合物2a和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入3.0mg(0.02mmol)1,8-二氮杂二环十一碳-7-烯混匀进行环加成反应。该反应体系中,化合物1i与化合物2a的摩尔比为1:1.2,1,8-二氮杂二环十一碳-7-烯占化合物1i的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到48.8mg产品3ia,收率76%。
产品3ia的核磁氢谱、核磁碳谱和高分辨质谱结果如下:
HRMS(ESI)calcd for C
实施例13
将29.9mg(0.1mmol)化合物1j,41.2mg(0.12mmol)化合物2a和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入3.0mg(0.02mmol)1,8-二氮杂二环十一碳-7-烯混匀进行环加成反应。该反应体系中,化合物1j与化合物2a的摩尔比为1:1.2,1,8-二氮杂二环十一碳-7-烯占化合物1j的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到46.9mg产品3ja,收率73%。
产品3ja的核磁氢谱、核磁碳谱和高分辨质谱结果如下:
HRMS(ESI)calcd for C
实施例14
将31.5mg(0.1mmol)化合物1k,41.2mg(0.12mmol)化合物2a和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入3.0mg(0.02mmol)1,8-二氮杂二环十一碳-7-烯混匀进行环加成反应。该反应体系中,化合物1k与化合物2a的摩尔比为1:1.2,1,8-二氮杂二环十一碳-7-烯占化合物1k的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到50.7mg产品3ka,收率77%。
产品3ka的核磁氢谱、核磁碳谱和高分辨质谱结果如下:
HRMS(ESI)calcd for C
实施例15
将33.5mg(0.1mmol)化合物1l,41.2mg(0.12mmol)化合物2a和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入3.0mg(0.02mmol)1,8-二氮杂二环十一碳-7-烯混匀进行环加成反应。该反应体系中,化合物1l与化合物2a的摩尔比为1:1.2,1,8-二氮杂二环十一碳-7-烯占化合物1l的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到48.8mg产品3la,收率72%。
产品3la的核磁氢谱、核磁碳谱和高分辨质谱结果如下:
HRMS(ESI)calcd for C
实施例16
将29.1mg(0.1mmol)化合物1m,41.2mg(0.12mmol)化合物2a和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入3.0mg(0.02mmol)1,8-二氮杂二环十一碳-7-烯混匀进行环加成反应。该反应体系中,化合物1m与化合物2a的摩尔比为1:1.2,1,8-二氮杂二环十一碳-7-烯占化合物1m的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到46.9mg产品3ma,收率74%。
产品3ma的核磁氢谱、核磁碳谱和高分辨质谱结果如下:
HRMS(ESI)calcd for C
实施例17
将26.9mg(0.1mmol)化合物1n,41.2mg(0.12mmol)化合物2a和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入3.0mg(0.02mmol)1,8-二氮杂二环十一碳-7-烯混匀进行环加成反应。该反应体系中,化合物1n与化合物2a的摩尔比为1:1.2,1,8-二氮杂二环十一碳-7-烯占化合物1n的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到52.1mg产品3na,收率85%。
产品3na的核磁氢谱、核磁碳谱和高分辨质谱结果如下:
HRMS(ESI)calcd for C
实施例18
将28.5mg(0.1mmol)化合物1a,33.9mg(0.12mmol)化合物2b和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入3.0mg(0.02mmol)1,8-二氮杂二环十一碳-7-烯混匀进行环加成反应。该反应体系中,化合物1a与化合物2b的摩尔比为1:1.2,1,8-二氮杂二环十一碳-7-烯占化合物1a的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到49.9mg产品3ab,收率88%。
产品3ab的核磁氢谱、核磁碳谱和高分辨质谱结果如下:
HRMS(ESI)calcd for C
实施例19
将28.5mg(0.1mmol)化合物1a,35.8mg(0.12mmol)化合物2c和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入3.0mg(0.02mmol)1,8-二氮杂二环十一碳-7-烯混匀进行环加成反应。该反应体系中,化合物1a与化合物2c的摩尔比为1:1.2,1,8-二氮杂二环十一碳-7-烯占化合物1a的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到45.6mg产品3ac,收率78%。
产品3ac的核磁氢谱、核磁碳谱和高分辨质谱结果如下:
HRMS(ESI)calcd for C
实施例20
将28.5mg(0.1mmol)化合物1a,35.8mg(0.12mmol)化合物2d和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入3.0mg(0.02mmol)1,8-二氮杂二环十一碳-7-烯混匀进行环加成反应。该反应体系中,化合物1a与化合物2d的摩尔比为1:1.2,1,8-二氮杂二环十一碳-7-烯占化合物1a的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到49.1mg产品3ad,收率84%。
产品3ad的核磁氢谱、核磁碳谱和高分辨质谱结果如下:
HRMS(ESI)calcd for C
实施例21
将28.5mg(0.1mmol)化合物1a,41.2mg(0.12mmol)化合物2e和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入3.0mg(0.02mmol)1,8-二氮杂二环十一碳-7-烯混匀进行环加成反应。该反应体系中,化合物1a与化合物2e的摩尔比为1:1.2,1,8-二氮杂二环十一碳-7-烯占化合物1a的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到54.0mg产品3ae,收率86%。
产品3ae的核磁氢谱、核磁碳谱和高分辨质谱结果如下:
HRMS(ESI)calcd for C
实施例22
将28.5mg(0.1mmol)化合物1a,33.4mg(0.12mmol)化合物2f和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入3.0mg(0.02mmol)1,8-二氮杂二环十一碳-7-烯混匀进行环加成反应。该反应体系中,化合物1a与化合物2f的摩尔比为1:1.2,1,8-二氮杂二环十一碳-7-烯占化合物1a的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到49.0mg产品3af,收率87%。
产品3af的核磁氢谱、核磁碳谱和高分辨质谱结果如下:
HRMS(ESI)calcd for C
实施例23
将28.5mg(0.1mmol)化合物1a,35.3mg(0.12mmol)化合物2g和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入3.0mg(0.02mmol)1,8-二氮杂二环十一碳-7-烯混匀进行环加成反应。该反应体系中,化合物1a与化合物2g的摩尔比为1:1.2,1,8-二氮杂二环十一碳-7-烯占化合物1a的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到54.5mg产品3ag,收率94%。
产品3ag的核磁氢谱、核磁碳谱和高分辨质谱结果如下:
HRMS(ESI)calcd for C
实施例24
将28.5mg(0.1mmol)化合物1a,31.7mg(0.12mmol)化合物2h和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入3.0mg(0.02mmol)1,8-二氮杂二环十一碳-7-烯混匀进行环加成反应。该反应体系中,化合物1a与化合物2h的摩尔比为1:1.2,1,8-二氮杂二环十一碳-7-烯占化合物1a的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到46.7mg产品3ah,收率83%。
产品3ah的核磁氢谱、核磁碳谱和高分辨质谱结果如下:
HRMS(ESI)calcd for C
实施例25
将28.5mg(0.1mmol)化合物1a,33.9mg(0.12mmol)化合物2i和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入3.0mg(0.02mmol)1,8-二氮杂二环十一碳-7-烯混匀进行环加成反应。该反应体系中,化合物1a与化合物2i的摩尔比为1:1.2,1,8-二氮杂二环十一碳-7-烯占化合物1a的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到51.1mg产品3ai,收率90%。
产品3ai的核磁氢谱、核磁碳谱和高分辨质谱结果如下:
HRMS(ESI)calcd for C
实施例26
将28.5mg(0.1mmol)化合物1a,33.4mg(0.12mmol)化合物2j和1mL四氢呋喃投入到干燥的15mL的史莱克管中,加入3.0mg(0.02mmol)1,8-二氮杂二环十一碳-7-烯混匀进行环加成反应。该反应体系中,化合物1a与化合物2j的摩尔比为1:1.2,1,8-二氮杂二环十一碳-7-烯占化合物1a的摩尔百分含量为20%,25℃下搅拌12小时,用旋转蒸发仪浓缩,经硅胶柱过柱纯化(二氯甲烷:石油醚=2:1,v/v),得到45.1mg产品3aj,收率80%。
产品3ai的核磁氢谱、核磁碳谱和高分辨质谱结果如下:
经过初步验证,本发明合成的化合物具有抗植物病原菌活性,具体的抑菌范围以及抑菌能力有待进一步研究。
- 一种环戊烷并香豆素类化合物及其制备方法
- 一种含有香豆素结构的木脂素类化合物及其制备方法与应用