用于合成含硫醚肽的方法
文献发布时间:2023-06-19 18:35:48
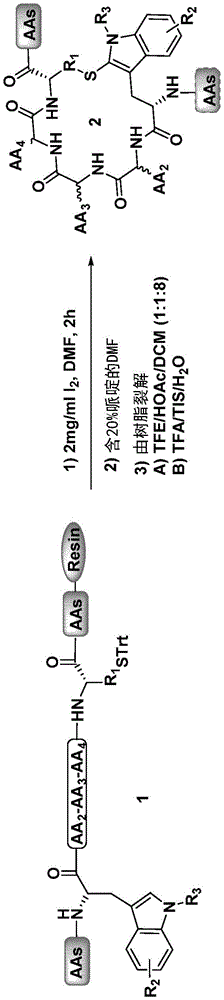
本发明涉及在碘处理下固相肽合成中在色氨酸与半胱氨酸之间形成硫桥的方法。本发明还涉及所述方法得到的化合物。
α-鹅膏蕈碱作为一种缓慢作用的毒素(LD
多年来已对氨基酸氨基羧乙基硫色氨酸的合成进行了探索,并且对于其形成有三种基本合成方法。氨基羧乙基硫色氨酸的早期合成包括使吲哚与硫基氯反应(Anderson,A.A.Shelat,R.K.Guy,J Org Chem 2005,70,4578-4584)。尽管该方法已被广泛使用,但是与作为获得氨基羧乙基硫色氨酸部分的额外合成方法的Savige-Fontana反应(Savige等人,J.Chem.Soc.,Chem.Commun.1976,600-601)相比,所涉及的费力的保护和脱保护策略使其不受欢迎。使用Savige-Fontana反应,进行鹅膏蕈碱的各种衍生物的初始构效关系以及α-鹅膏蕈碱的首次全合成(Zanotti等人,Chem-Eur J 2001,7,1479-1485;Matinkhoo等人,J Am Chem Soc 2018,140,6513-651)。
此前,碘介导的未保护的Trp侧链和三苯甲基保护的Cys的硫代反应已经在一些模型肽的实例中进行了描述(Sieber等人,Helv Chim Acta 1980,63,2358-2363)。该方法已被应用于鬼笔环肽及衍生物的合成(Schuresko等人,Angew Chem Int Edit 2007,46,3547-3549;Yao等人,Chem-Eur J 2019,25,8030-8034),但未对鹅膏蕈碱型肽及其衍生的序列进行描述,因为肽序列的不同长度、次序和分子内氢键会形成不同的结果和收率。
基于上文所提及的现有技术,本发明的目的在于提供在固相肽合成中形成氨基羧乙基硫色氨酸型硫桥的手段和方法,以用于合成环肽,特别是合成鹅膏毒素及衍生物。该目的通过本说明书的独立权利要求的主题来获得。
发明概述
本发明涉及用于制备式(I)的化合物的方法,
附图说明
图1单环五肽的固相肽合成。
图2单环肽的固相肽合成。
图3单环肽的固相肽合成。
图4(A)α-膏蕈碱以及A环和B环的结构,(B)抗体-毒素-缀合物的示意性实例。
图5单环五肽的溶液相肽合成。
图6Fmoc-L-Trp(6-OBn)-OH的合成。a)POCl
图7Cbz-L-Trp(Boc)(6OBn)-OMe的合成。A)Cbz-2-膦酰甘氨酸-甲基二甲酯,DBU,DCM,0℃至室温,16小时;b)H
图8Boc-D-Trp(Boc)(6-OBn)-OMe的合成,H
图9显示不同鹅膏毒素的结构式。粗体形式的数字(1至8)指定形成鹅膏毒素的八个氨基酸的标准编号。还显示了氨基酸1、3和4中原子的标准编号(分别为希腊字母α至γ,希腊字母α至δ,以及1′至7′的数字)。A环和B环被标记。
氨基酸序列从氨基端到羧基端给出。序列位置的大写字母是指单字母代码中的L-氨基酸(Stryer,Biochemistry,第3版,第21页)。氨基酸序列位置的小写字母是指相应的D-或(2R)-氨基酸。
在本说明书的上下文中,术语“保护基”涉及与官能团共价连接的部分(特别是本文所讨论的分子的羧酸部分、氨基部分或羟基部分),该部分可选择性地与官能团连接并选择性地移除,而不影响保护基所连接的分子的碳主链的完整性或手性取向,也不裂解与分子连接的特定其它保护基。
在本说明书的上下文中,术语“脱保护剂”涉及能够裂解某些保护基的物质。技术人员能够根据保护基来选择脱保护剂。可使保护基裂解的条件设立脱保护剂,例如,如果保护基可在酸性条件下裂解,则脱保护剂是酸。
现代保护基化学的全面综述,特别是在其涉及本文所公开的化合物时,可获得于Peter G.M.Wuts,Greene′s Protective Groups in Organic Synthesis,第5版,Wiley2014。
US 6693178 B2-“Protecting groups useful in the synthesis ofpolysaccharides,natural products,and combinatorial libraries”和US 20160024143A1-″Deprotection method”通过援引加入本文。
本文遵循有机化学的标准惯例,据此式中的未指定位置被认为是饱和碳。
氨基酸残基序列从氨基端到羧基端给出。序列位置的大写字母是指单字母代码中的L-氨基酸(Stryer,Biochemistry,第3版,第21页)。氨基酸序列位置的小写字母是指相应的D-或(2R)-氨基酸。序列以从氨基端到羧基端的方向从左到右书写。根据标准命名法,氨基酸残基序列用如下所指示的三字母或单字母代码命名:丙氨酸(Ala,A)、精氨酸(Arg,R)、天冬酰胺(Asn,N)、天冬氨酸(Asp,D)、半胱氨酸(Cys,C)、谷氨酰胺(Gln,Q)、谷氨酸(Glu,E)、甘氨酸(Gly,G)、组氨酸(His,H)、异亮氨酸(Ile,I)、亮氨酸(Leu,L)、赖氨酸(Lys,K)、甲硫氨酸(Met,M)、苯丙氨酸(Phe,F)、脯氨酸(Pro,P)、丝氨酸(Ser,S)、苏氨酸(Thr,T)、色氨酸(Trp,W)、酪氨酸(Tyr,Y)和缬氨酸(Val,V)。如果未指明,则氨基酸是L-氨基酸。
当在本文中以最狭隘的意义使用时,术语未取代的C
当在化学式的语境中使用时,可以使用以下缩写:Me是甲基CH
在本说明书的上下文中,术语杂芳基涉及包含至少一个杂原子(例如N、O、S),特别是一个或数个氮、氧和/或硫原子的环状芳族C
术语取代的杂芳基在其最广泛的意义上是指如上在最广泛的意义上所定义的杂芳基,其共价连接至不是碳或氢的原子,特别是选自N、O、F、B、Si、P、S、Cl、Br和I的原子,其本身如果适用可以连接至该基团的一个或数个其它原子,或氢,或者不饱和或饱和烃(在其最广泛的意义上为烷基或芳基)。在最狭隘的意义上,取代的杂芳基是指如上在最广泛的意义上所定义的杂芳基,其在一个或数个碳原子上被选自以下的基团取代:胺NH
本发明的方法涉及固相肽合成,其中在氧化条件下,特别是在碘处理下形成硫桥。
本发明的第一方面涉及用于制备式(I)的化合物的方法,
其中
在反应步骤(a)中,使式(II)的化合物
与碘(I
其中
-Z选自SH、S-三苯甲基(STrt)、S-乙酰氨基甲基(SAcm)、S-二苯甲基(SDpm)、S-单甲氧基三苯甲基(SMmt)和S-叔丁基(S
-Y是未取代的或羟基-、卤素-、卤代碳-、炔基-、烯烃.、氰基-保护的羧酸酯和/或羧基酰胺取代的吲哚,特别是未取代的或羟基-、卤素-、卤代碳-、炔基-、烯烃-、氰基-保护的羧酸酯和/或羧基酰胺取代的吲哚;
-L
-L
-B是α-氨基酸、β-氨基酸主链、α-甲基氨基酸,特别地B是α-氨基酸主链(NH-CHR-C=O,其中R是L
-每个A独立地选自呈L-或D-构象的蛋白原性和非蛋白原性α-氨基酸或β-氨基酸,特别地每个A独立地选自呈L-或D-构象的蛋白原性和非蛋白原性α-氨基酸;
-n是选自2、3和4的整数;
-条件是环
由14至26个键,特别是15至21个键,更特别是18个键组成;
-每个C和D独立地选自呈L-或D-构象的蛋白原性和非蛋白原性α-氨基酸,其中式(II)的C或D与树脂连接或者具有受保护的N端;
-m和k独立地选自0至4的整数,特别地其中m和k的总和为0至4的整数;
-X是(-(-吲哚-S-)或(-S-吲哚-)-),其中吲哚是未取代的或羟基-、卤素-、卤代碳-、炔基-、烯烃-、氰基-保护的羧酸酯和/或羧基酰胺取代的吲哚,其中任选地x的硫原子可以随后被氧化。
所述化合物可由式(Ia)或式(Ib)表示,
特别地,条件是从左至右的方向是从N端至C端或从C端至N端。
如果没有不同说明,与碘的反应如“I
固相肽合成在固体载体上进行。固体载体由用反应性基团官能化的小聚合物树脂珠组成。本发明的硫桥形成可以在肽仍然与树脂连接时或者在从树脂上裂解后进行。如果在裂解后进行,肽的N端用氨基保护基保护。
在某些实施方案中,树脂载量为约0.3mmol/g。在较高的树脂载量下,会促进副反应。
α-L-氨基酸主链具有式
在某些实施方案中,式(II)的C或D与树脂连接,并与碘(I
在某些实施方案中,式(II)的C或D具有受保护的N端,并与碘(I
如果线性肽与树脂偶联,则2当量的碘可以加速固相上的反应。固体载体可以使得与过量的试剂和溶剂一同起作用。因此,就副反应而言,两当量不是关键的。
相比之下,对于溶液中的肽,仅使用一当量的碘来防止副反应。
在某些实施方案中,反应步骤(a)在极性溶剂中进行。在某些实施方案中,反应步骤(a)在MeOH、DCM、NMP(N-甲基-2-吡咯烷酮)或DMF中进行。在某些实施方案中,反应步骤(a)在极性非质子溶剂中进行。在某些实施方案中,反应步骤(a)在DMF中进行。在某些实施方案中,反应步骤(a)在温和酸性条件下(如在DCM中含有TFA(低浓度,如1%)、TFE(三氟乙醇)、AcOH或HFIP(六氟异丙醇)的溶剂混合物)进行。
在某些实施方案中,碘以1-4mg/ml,特别是2mg/ml的浓度使用。
在某些实施方案中,n是3。
在某些实施方案中,A独立地选自呈L-或D-构象的蛋白原性或非蛋白原性α-氨基酸。在某些实施方案中,A独立地选自未取代的或羟基取代的Gly、Ala、Ile、Leu、Val、Pro、Phe、Lys、Arg、His、D-Pro、D-Ala、L-炔丙基-Gly、Aib(式
光氨基酸包含式
叠氮氨基酸包含叠氮基(-N
炔基氨基酸包含式
在某些实施方案中,C和D独立地选自呈L-或D-构象的蛋白原性或非蛋白原性α-氨基酸。在某些实施方案中,C和D独立地选自Gly、Ala、Ile、Leu、Val、Pro、Phe、Lys、Ser、Cys、Arg、His、Asp、Asn、Gln、Glu、Hyp、L-哌啶甲酸(3105-95-1)、L-氮杂环丁烷-2-甲酸(2133-34-8)、(S)-二氢吲哚-2-甲酸(79815-20-6)、L-4-噻唑烷甲酸(34592-47-7)、反式-4-Hyp(式
羟基化氨基酸包含至少一个OH基团。
在某些实施方案中,Y的吲哚未被取代或者被一个、两个、三个或四个选自羟基、卤素、CN和氟化碳(CF
如果Y包含羟基,则该基团可以脱保护或者用羟基保护基保护,优选地保护。
在某些实施方案中,Y的吲哚是未取代的吲哚。在某些实施方案中,吲哚经由其3-位
在某些实施方案中,吲哚经由其2-位连接至硫原子。
在某些实施方案中,树脂是酸不稳定树脂。在某些实施方案中,树脂是2-氯三苯甲基树脂、Rink酰胺树脂、1,3-二氢-2H-吡喃-2-基-甲氧基甲基树脂(THP树脂)或王氏树脂。
在某些实施方案中,Z的硫原子是氧化的。在某些实施方案中,Z的硫原子通过如下方式氧化:
i.使用锰离子,特别地,使化合物与式(V)的化合物,
以及与Mn(OTf)
ii使用PPO、过氧化二苯甲酰、过氧苯甲酸叔丁酯或过氧化月桂酰;或mCPBA;
iii.使用碘和氧气。
本发明的第二方面涉及用于制备式(VI)的化合物的方法,
其中在反应步骤(a)中,使式(VII)的化合物
其中
·R
·R
·R
·R
与H
对于R
对于R
在某些实施方案中,在反应步骤(b)中,使式(VIII)的化合物
其中
·R
与受保护的2-膦酰甘氨酸-甲基二甲酯在碱性条件下反应,特别地与Boc-2-膦酰甘氨酸-甲基二甲酯、Cbz-2-膦酰甘氨酸-苄基二甲酯或Cbz-2-膦酰甘氨酸-甲基二甲酯在碱性条件下反应,更特别地碱选自DBU(1,8-二氮杂双环(5.4.0)十一碳-7-烯)和四甲基胍(CAS 80-70-6),
以得到以(VII)为特征的化合物。
本发明的第三方面涉及式(IIIa)和(IIIb)的化合物,
或者式(IVa)、(IVb)、(IVc)、(IVd)、(IVe)、(IVf)、(IVg)或(IVh)的化合物,
其中
-X在式[(IVa)、(IVb)、(IVc)和(IVd)]中为式Q-R,或在式[(IVe)、(IVf)、(IVg)和(IVh)]中为R-Q,
-Q是未取代的或CF
-R是S、SO或SO
-L
-L
-B是α-氨基酸主链;
-AA
-AA
-AA
-AA
-AA
-AA
特别地条件是在式(IIIa)中,AA
并且在式(IVa)、(IVb)、(IVc)、(IVd)、(IVe)、(IVf)、(IVg)或(IVh)中,C端氨基酸任选地与树脂连接;
特别地,条件是从左至右的方向是从N端至C端或从C端至N端。
在根据本发明的化合物的具体实施方案中,所述化合物不由以下组合构成:
·Q是未取代的或烷基-、O-烷基-、羟基-和/或卤素-取代的吲哚;
·R是S、SO或SO
·L
·L
·AA
·AA
·AA
·AA
·AA
·AA
或者
·Q是未取代的或烷基-、O-烷基-、羟基-和/或卤素-取代的吲哚;
·R是S、SO或SO
·L
·L
·AA
·AA
·AA
·AA
·AA
或者
·Q是未取代的或烷基-、O-烷基-、羟基-和/或卤素-取代的吲哚;
·R是S、SO或SO
·L
·L
·AA
·AA
·AA
·AA
·AA
·AA
在某些实施方案中,吲哚经由其3-位连接至连接基L
在一些实施方案中,本发明提供可通过如本文所公开的本发明方法获得的化合物。
在一些实施方案中,可通过如上所公开的本发明方法获得的化合物用于制备抗体药物缀合物(ADC)。
根据一个实施方案,本发明涉及可通过如本文所公开的本发明方法获得的化合物在制备抗体-药物缀合物中的用途,其中所述化合物选自包含以下的化合物组:
根据一个实施方案,本发明涉及如上所公开的本发明化合物作为ADC有效载荷的用途。如本发明中所用,术语“有效载荷”是指生物活性细胞毒性(抗癌)药物,例如可通过本发明方法获得的化合物,例如本发明的化合物7a-7r,其缀合至(i)抗体,优选单克隆抗体,(ii)其抗原结合片段,优选可变域(Fv)、Fab片段或F(ab)2片段,(iii)其抗原结合衍生物,优选单链Fv(scFv),以及(iv)抗体样蛋白。
如本文所用,术语“抗体”应当指由免疫球蛋白基因或免疫球蛋白基因片段或由其衍生的cDNA编码的一条或多条多肽链组成的蛋白质。所述免疫球蛋白基因包括轻链κ、λ以及重链α、δ、ε、γ和μ恒定区基因,以及多种不同可变区基因中的任一种。
基本免疫球蛋白(抗体)结构单元通常是由两对相同的多肽链即轻链(L,分子量为约25kDa)和重链(H,分子量为约50-70kDa)构成的四聚体。每条重链由重链可变区(缩写为VH或VH)和重链恒定区(缩写为CH或CH)构成。重链恒定区由三个域组成,即CH1、CH2和CH3。每条轻链含有轻链可变区(缩写为VL或VL)和轻链恒定区(缩写为CL或CL)。VH和VL区可进一步细分为超变区,也称为互补决定区(CDR),散布有称为框架区(FR)的更保守区域。每个VH和VL区由从氨基端到羧基端以FR1、CDR1、FR2、CDR2、FR3、CDR3、FR4的次序布置的三个CDR和四个FR构成。重链和轻链的可变区形成与抗原相互作用的结合域。
CDR对于抗体或其抗原结合部分的结合最为重要。只要是抗原结合所需的三维结构得到保留,则FR可被其它序列替代。构建体的结构变化最常导致与抗原的充分结合丧失。
术语(单克隆)抗体的“抗原结合片段”是指保留以其天然形式特异性结合其抗原的能力的抗体的一个或多个片段。抗体的抗原结合部分的实例包括Fab片段、由VL、VH、CL和CH1域组成的单价片段、F(ab’)2片段、包含在铰链区处经二硫桥连接的两个Fab片段的二价片段、由VH和CH1域组成的Fd片段、由抗体的单臂的VL和VH域组成的Fv片段,以及由VH域和分离的互补决定区(CDR)组成的dAb片段。
根据本发明的抗体或其抗体片段或抗体衍生物可以是单克隆抗体。如本文所用,术语“单克隆抗体”(“mAb”)是指针对特定表位有单一结合特异性和亲和力的抗体分子的制剂,代表同质抗体群体,即,由全免疫球蛋白或其片段或衍生物组成的同质群体。优选地,这样的抗体选自IgG、IgD、IgE、IgA和/或IgM、或其片段或衍生物,优选地本发明的单克隆抗体为IgG同种型,例如IgG1或IgG4,更优选地为IgG1同种型。
如本文所用,术语“片段”或“抗原结合片段”应当指保留靶结合能力的这样的抗体的片段,例如CDR(互补决定区)、高变区、可变域(Fv)、IgG重链(由VH、CH1、铰链区、CH2和CH3区组成)、IgG轻链(由VL和CL区组成),和/或Fab和/或F(ab)2。
如本文所用,术语“衍生物”或“抗原结合衍生物”应指在结构上不同于普遍抗体概念,但仍与普遍抗体概念具有一些结构关系的蛋白质构建体,例如scFv、Fab和/或F(ab)2,以及双特异性、三特异性或更高特异性的抗体构建体,所有这些都具有与本发明的单克隆抗体大致相同的靶结合特异性。
技术人员已知的其他抗体衍生物是双抗体、骆驼抗体、域抗体、具有由scFv、IgA组成的两条链的二价同源二聚体(由J链和分泌片连接的两个IgG结构)、鲨鱼抗体(IgNAR)、由新世界灵长类框架加上非新世界灵长类CDR组成的抗体、包含CH3+VL+VH的二聚化构建体、包含CDR的其它支架蛋白形式,以及抗体缀合物(例如,与药物、毒素、细胞因子、适体、核酸如脱氧核糖核酸(DNA)或核糖核酸(RNA)、治疗性多肽、放射性同位素或标记连接的抗体或其片段或衍生物)。
如本文中所使用,术语“抗体样蛋白”是指经工程改造(例如,通过诱变Ig环)后与靶向分子特异性结合的蛋白。通常,此类抗体样蛋白包含至少一个在两端与蛋白骨架连接的可变肽环。该双结构约束大大增加了抗体样蛋白的结合亲和力,使其水平与抗体的相当。所述可变肽环的长度通常由10-20个氨基酸组成。所述支架蛋白可以是任何具有良好溶解度特性的蛋白。优选地,所述支架蛋白为小的球状蛋白。抗体样蛋白包括但不限于affilin蛋白、亲和体、抗运载蛋白(anti-calin)和设计的锚蛋白重复蛋白(Binz等人,2005)。抗体样蛋白可以来源于大型的突变文库,例如通过从大型噬菌体展示文库中淘选,并且可以类似于常规抗体进行分离。同时,通过对球状蛋白中表面暴露残基的组合诱变,可以获得抗体样结合蛋白。
根据一个实施方案,如上所公开的本发明的化合物可以例如直接经由键或经由连接基,优选地经由连接基与(i)抗体,优选单克隆抗体,(ii)其抗原结合片段,优选可变域(Fv)、Fab片段或F(ab)2片段,(iii)其抗原结合衍生物,优选单链Fv(scFv),以及(iv)抗体样蛋白中的任一种偶联。如本发明的上下文中所用,术语“连接基”是指使两个组分连接的结构,每个组分与连接基的一端连接。
在特定实施方案中,连接基增加两个组分之间的距离,并且减轻这些组分之间(如在本发明的情况下,抗体与本发明的化合物之间)的空间干扰。在特定实施方案中,所述连接基在其主链中具有1至30个原子(例如,1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、或30个原子)的连续链,即,连接基的长度被定义为如由本发明化合物部分与以下之间的原子或键的数量所测量的最短连接:(i)抗体,优选单克隆抗体,(ii)其抗原结合片段,优选可变域(Fv)、Fab片段或F(ab)2片段,(iii)其抗原结合衍生物,优选地单链Fv(scFv),或(iv)抗体样蛋白,其中连接基主链的一侧已经与本发明的化合物反应,而另一侧可用于反应,或者已经与例如抗体反应。在本发明的上下文中,连接基是例如任选取代的C
在特定实施方案中,连接基可以是1至20个独立地选自C、O、N和S的原子,特别是2至18个原子,更特别是5至16个原子,并甚至更特别是6至15个原子的直链。在特定实施方案中,直链中至少60%的原子是C原子。在特定实施方案中,直链中的原子经由单键连接。
在特定实施方案中,连接基可以是亚烷基、亚杂烷基、亚烯基、亚杂烯基、亚炔基、亚杂炔基、亚环烷基、亚杂环烷基、亚芳基、亚杂芳基、亚芳烷基或包含1至4个选自N、O和S的杂原子的亚杂芳烷基,其中所述连接基任选地被取代。
术语“亚烷基”是指具有1至20个碳原子的二价直链饱和烃基,包括具有1至10个碳原子的基团。在某些实施方案中,亚烷基可以是低级亚烷基。术语“低级亚烷基”是指具有1至6个碳原子,并且在某些实施方案中1至5个或1至4个碳原子的亚烷基。亚烷基的实例包括但不限于亚甲基(-CH
术语“亚烯基”是指具有2至20个碳原子的二价直链基团,其中至少一个碳-碳键是双键,而其它键可以是单键或另一些双键。本文中的术语“亚炔基”是指具有2至20个碳原子的基团,其中至少一个碳-碳键是三键,而其它键可以是单键、双键或另一些三键。亚烯基的实例包括亚乙烯基(-CH=CH-)、1-亚丙烯基、2-亚丙烯基、1-亚丁烯基、2-亚丁烯基、3-亚丁烯基等。亚炔基的实例包括亚乙炔基、1-亚丙炔基、2-亚丙炔基等。
如本文所用,“亚环烷基”旨在指作为任何稳定的单环或多环体系的一部分的二价环,其中这样的环具有3至12个碳原子,但不含杂原子,并且其中这样的环是完全饱和的,并且术语“亚环烯基”旨在指作为任何稳定的单环或多环体系的一部分的二价环,其中这样的环具有3至12个碳原子,但不含杂原子,并且其中这样的环是至少部分不饱和的(但任何亚芳基环除外)。亚环烷基的实例包括但不限于亚环丙基、亚环丁基、亚环戊基、亚环己基和亚环庚基。环亚烯基的实例包括但不限于亚环戊烯基和亚环己烯基。
如本文所用,术语“亚杂环烷基”和“亚杂环烯基”旨在指作为任何稳定的单环或多环环系的一部分的二价环,其中这样的环具有3至约12个原子,并且其中这样的环由碳原子和至少一个杂原子(特别是至少一个独立地选自N、O和S的杂原子)组成,其中亚杂环烷基是指完全饱和的环,并且亚杂环烯基是指至少部分不饱和的环(但任何亚芳基或亚杂芳基环除外)。
术语“亚芳基”旨在意指作为任何稳定的单环或多环体系的一部分的二价环或环系,其中这样的环或环系具有3至20个碳原子,但不具有杂原子,该环或环系由如通过“4n+2”π电子规则所定义的芳族部分组成,包括亚苯基。
如本文所用,术语“亚杂芳基”是指作为任何稳定的单或多环体系的一部分的二价环或环系,其中这样的环或环系具有3至20个原子,该环或环系由如通过“4n+2”π电子规则所定义的芳族部分组成并且含有碳原子以及一个或多个氮、硫和/或氧杂原子。
在本发明的上下文中,术语“取代的”旨在指示在连接基的主链中存在的一个或多个氢被指定基团的选择所替代,条件是不超过所指定原子的正常化合价或被取代的基团的适当原子的化合价,并且取代产生了稳定的化合物。术语“任选取代的”旨在意指连接基是未取代的或者如本文所定义被一个或多个如本文所定义的取代基取代。当取代基是酮基(或氧代,即=O)、硫基或亚氨基等时,则连接基主链原子上的两个氢被替代。示例性取代基包括例如烷基、烯基、炔基、环烷基、杂环烷基、芳基、杂芳基、芳烷基、杂芳烷基、酰基、芳酰基、杂芳酰基、羧基、烷氧基、芳氧基、酰氧基、芳酰氧基、杂芳酰氧基、烷氧羰基、卤素、(硫代)酯、氰基、磷酰基、氨基、亚氨基、(硫代)酰胺基、巯氢基、烷硫基、酰硫基、磺酰基、硫酸酯、磺酸酯、氨磺酰基、磺酰胺基、硝基、叠氮基、卤代烷基(包括全氟烷基(如三氟甲基))、卤代烷氧基、烷基硫烷基、烷基亚磺酰基、烷基磺酰基、烷基磺酰基氨基、芳基磺酰氨基、磷酰基、磷酸酯、膦酸酯、亚膦酸酯、烷基羧基、烷基羧基酰胺、氧代、羟基、巯基、氨基(任选地单或二取代的,例如被烷基、芳基或杂芳基)、亚氨基、甲酰胺、氨基甲酰基(任选地单或二取代的,例如被烷基、芳基或杂芳基)、脒基、氨基磺酰基、酰基氨基、芳酰基氨基、(硫代)脲基、(芳基硫代)脲基、烷基(硫代)脲基、环烷基(硫代)脲基、芳氧基、芳烷氧基或-O(CH
在特定实施方案中,连接基L包含选自以下部分之一的部分:二硫化物(-S-S-)、醚(-O-)、硫醚(-S-)、胺(-NH-)、酯(-O-C(=O)-或-C(=O)-O-)、甲酰胺(-NH-C(=O)-或-C(=O)-NH-)、氨基甲酸酯(-NH-C(=O)-O-或-O-C(=O)-NH-)和脲部分(-NH-C(=O)-NH-)。
在本发明的特定实施方案中,连接基L包含m个选自以下列表的基团:亚烷基、亚烯基、亚炔基、亚环烷基、亚杂烷基、亚杂烯基、亚杂炔基、亚杂环烷基、亚芳基、亚杂芳基、亚芳烷基和亚杂芳烷基,其中各基团可任选地是独立取代的,连接基还包含n个独立地选自以下部分之一的部分:二硫化物(-S-S-)、醚(-O-)、硫醚(-S-)、胺(-NH-)、酯(-O-C(=O)-或-C(=O)-O-)、甲酰胺(-NH-C(=O)-或-C(=O)-NH-)、氨基甲酸酯(-NH-C(=O)-O-或-O-C(=O)-NH-)和脲部分(-NH-C(=O)-NH-),其中m=n+1。在特定实施方案中,m是2且n是1,或者m是3且n是2。在特定实施方案中,连接基包含2或3个未取代的亚烷基,以及1或2个分别连接未取代的亚烷基的二硫化物、醚、硫醚、胺、酯、甲酰胺、氨基甲酸酯或脲部分。
在特定实施方案中,直链中的C原子独立地为任选取代的亚甲基(CH
所述连接基可以例如缀合到如本文所公开的本发明的化合物的羟脯氨酰残基(Hyp),或者本发明的化合物的DHIle(二羟基-异亮氨酸)残基。连接基与本发明化合物的Hyp残基的缀合可以例如根据如EP3735987A1(将其内容援引加入)中所公开的方法进行,连接基与本发明化合物的DHIle(二羟基-异亮氨酸)的缀合可以根据WO2020234461 A1(将其内容援引加入)中所公开的方法进行。
根据一些实施方案,与如本文所公开的本发明化合物缀合的连接基可以是不可裂解的(稳定的)或可裂解的连接基。在本发明的上下文中,术语“稳定的连接基”是指(i)在存在酶,特别是溶酶体肽酶(如组织蛋白酶B)的情况下,以及(ii)在细胞内还原环境中稳定的连接基。
根据一个实施方案,稳定的连接基不含有(i)酶可裂解亚结构,特别是不含可由组织蛋白酶B裂解的二肽序列,和/或(ii)二硫化物基团。在具体此类实施方案中,连接基具有多至12个原子,特别地2至10,更特别地4至9,并且最特别地6至8个原子的长度。
在本发明的上下文中,“可裂解连接基”被理解为包含至少一个裂解位点。如本文所用,术语“裂解位点”应当指在特定条件下,在限定位置处易受特定裂解影响的部分。所述条件是例如特定酶或特定机体或细胞区室中的还原性环境。根据本发明的酶可裂解部分也可称为“可被酶裂解”。连接基的酶促裂解导致胞内释放与如本文所公开的靶向部分或抗体缀合的毒素载物、或其内化后的代谢物(参见Dubowchik等人,Bioconjug Chem.13(2002)855-69)。
所述可裂解连接基可以选自酶促可裂解连接基,优选地蛋白酶促可裂解连接基,以及化学可裂解连接基,优选地包含二硫桥的连接基。
根据本发明的优选实施方案,所述裂解位点是包含两个或更多个氨基酸的酶促可裂解部分。优选地,所述酶促可裂解部分包含缬氨酸-丙氨酸(Val-Ala)、缬氨酸-瓜氨酸(Val-Cit)、缬氨酸-赖氨酸(Val-Lys)、缬氨酸-精氨酸(Val-Arg)二肽、苯丙氨酸-赖氨酸-甘氨酸-脯氨酸-亮氨酸-甘氨酸(Phe Lys Gly Pro Leu Gly)或丙氨酸-丙氨酸-脯氨酸-缬氨酸(Ala Ala Pro Val)肽或β-葡糖苷酸或β-半乳糖苷。
根据一些实施方案,所述裂解位点可以被选自半胱氨酸蛋白酶、金属蛋白酶、丝氨酸蛋白酶、苏氨酸蛋白酶和天冬氨酸蛋白酶的至少一种蛋白酶裂解。
半胱氨酸蛋白酶(也称为硫醇蛋白酶)是具有共同的催化机制的蛋白酶,所述机制涉及催化三联体或二联体中的亲核半胱氨酸硫醇。
金属蛋白酶是其催化机理涉及金属的蛋白酶。大多数金属蛋白酶需要锌,但有些使用钴。金属离子经由三个配体与蛋白质配位。配位金属离子的配体可随组氨酸、谷氨酸、天冬氨酸、赖氨酸和精氨酸而变化。第四配位位置由不稳定的水分子占据。
丝氨酸蛋白酶是裂解蛋白质中的肽键的酶,丝氨酸在(酶的)活性位点充当亲核氨基酸。丝氨酸蛋白酶根据其结构分为两大类:胰凝乳蛋白酶样(胰蛋白酶样)或枯草杆菌蛋白酶样。
苏氨酸蛋白酶是在活性位点内携带苏氨酸(Thr)残基的蛋白水解酶家族。这类酶的原型成员是蛋白酶体的催化亚单位,然而,酰基转移酶趋同地演化出相同的活性位点几何结构和机理。
天冬氨酸蛋白酶是一种催化类型的蛋白酶,其使用与一个或多个天冬氨酸残基结合的活化水分子来催化其肽底物。通常,它们在活性位点中具有两个高度保守的天冬氨酸,并且在酸性pH下具有最佳活性。胃蛋白酶抑制剂抑制几乎所有已知的天冬氨酰蛋白酶。
在本发明的具体实施方案中,所述裂解位点被选自以下中的至少一种物质裂解:组织蛋白酶A或B、基质金属蛋白酶(MMP)、弹性蛋白酶、β-葡糖醛酸糖苷酶和β-半乳糖苷酶,优选组织蛋白酶B。
在特别优选的实施方案中,根据本发明的酶促可裂解的连接基包含选自Phe-Lys、Val-Lys、Phe-Ala、Val-Ala、Phe-Cit和Val-Cit的二肽,特别是其中可裂解的连接基进一步包含二肽和可通过本发明方法获得的本发明化合物之间的对氨基苄基(PAB)间隔基,例如上文公开的化合物7a、7b、7c、7d、7e、7g、7h、7i、7j、7l、7m、7n、7o、7p、7q、7r中的一种,通过PAB部分连接或缀合至本发明的化合物:
如本文所公开的根据本发明的连接基-化合物缀合物与抗体,优选单克隆抗体、其抗原结合片段如可变域(Fv)、Fab片段或F(ab)2片段、或其抗原结合衍生物的缀合可以通过缀合至反应性赖氨酸或半胱氨酸残基(参见例如Jain等人,Pharm Res(2015)32:3526-3540),优选通过缀合至反应性半胱氨酸残基来进行,以得到抗体-药物缀合物(ADC)。
根据一些实施方案,本发明化合物与反应性赖氨酸残基的偶联或缀合可以使用包含以下反应性基团之一的连接基进行:1)SPDB二硫化物,2)MCC(马来酰亚胺基甲基环己烷-1-羧酸酯),3)增加有带电极性基团的磺基-SPDB,以及4)腙。相应的缀合可例如如Peeters等人,J Immunol Methods.1989年6月2日;120(1):133-43,或Carlsson等人,BiochemJ.1978;173:723-37中所述进行。
根据优选的实施方案,如上所公开的本发明的连接基包含硫醇反应性基团,其选自溴乙酰胺、碘乙酰胺、甲基磺酰基苯并噻唑、4,6-二氯-1,3,5-三嗪-2-基氨基基团甲基-磺酰基苯基四唑或甲基磺酰基苯基噁二唑、吡啶-2-硫醇、5-硝基吡啶-2-硫醇、甲烷硫代磺酸酯或马来酰亚胺,优选马来酰亚胺(马来酰亚胺基残基)。使用基于马来酰亚胺基的缀合来与反应性半胱氨酸残基缀合可以例如如WO2016/142049中所公开进行。
根据一个实施方案,如上所公开的抗体-药物-缀合物包含经由如本文所公开的连接基与抗体的反应性赖氨酸残基或半胱氨酸残基偶联的约1至约10,优选约1、2至约4、5、6、7、8个如本文所公开的本发明的化合物。
根据一个优选的实施方案,如上所公开的根据本发明的连接基-化合物缀合物可以例如通过半胱氨酸缀合经由位点特异性缀合而缀合至包含根据EU编号系统的重链118Cys或重链256Cys的半胱氨酸工程化抗体(Edelman等人,Proc.Natl.Acad.Sci.USA;63(1969)),如WO2016/142049 A1中所公开,其内容通过援引加入本文。使用所述半胱氨酸工程化抗体可特别有利地获得包含如本文所公开的本发明化合物的抗体-药物缀合物,其具有约2的受控的药物与抗体比率(DAR)(例如,抗体的每条重链有一个连接基-化合物缀合物),其例如可导致与DAR较高的ADC相比更好的所述ADC的治疗指数,或者包含DAR为约1至约6、8、或10的ADC物质的混合物的ADC制剂。如本文所用,术语“治疗指数”是药物的相对安全性的定量量度,并且可以例如定义为TI=TD
根据一个实施方案,如上所公开的半胱氨酸工程化抗体可额外在其Fc区中包含氨基酸取代L234A、L235A (根据EU编号系统),如WO 2020/086776 A1中所公开,其内容通过援引加入本文。使用这种工程化抗体可例如有利于进一步改善抗体-药物缀合物的治疗指数(TI),所述抗体-药物缀合物包含可通过本发明方法获得的化合物,例如,如本文所公开的化合物7a、7b、7c、7d、7e、7g、7h、7i、7j、7l、7m、7n、7o、7p、7q、7r之一。
根据一些实施方案,本发明涉及包含可通过本发明方法获得的本发明化合物或包含如上所公开的本发明化合物的抗体-药物缀合物。例如,根据本发明的ADC可以包含约1、2、4至约6、8、10,或约2、4、6、8,或约2至约4,或约2种如本文所公开的本发明化合物7a、7b、7c、7d、7e、7g、7h、7i、7j、7l、7m、7n、7o、7p、7q、7r,其可以例如经由如本文所公开的稳定或可裂解连接基缀合至抗体,由此给定的ADC会不包含如上所公开的本发明化合物的混合物。
无论在本文何处将单个可分离特征(例如,氨基酸或氧化方法)的替代方案列为“实施方案”,应理解,此类替代方案均可自由组合,以形成本文所公开的本发明的离散实施方案。因此,氨基酸的任何替代实施方案可以与本文所提及的氧化方法的任何替代实施方案组合。
通过以下实施例和附图进一步说明本发明,从中可以得出进一步的实施方案和优点。这些实施例意在说明本发明而非限制其范围。
实施例
表1:通过碘环化的结合至固体载体的肽的氨基酸序列。在从树脂上裂解并通过制备型HPLC纯化后计算分离收率(参见图1)。
化合物2a:HPLC-MS:保留时间R
化合物2b:HPLC-MS:保留时间R
化合物2c:HPLC-MS:保留时间R
化合物2d:HPLC-MS:保留时间R
化合物2e:HPLC-MS:保留时间R
化合物2f:HPLC-MS:保留时间R
化合物2g:HPLC-MS:保留时间R
化合物2h:HPLC-MS:保留时间R
化合物2i:HPLC-MS:保留时间R
化合物2j:HPLC-MS:保留时间R
化合物2k:HPLC-MS:保留时间R
化合物2l:HPLC-MS:保留时间R
化合物2m:HPLC-MS:保留时间R
化合物2n:HPLC-MS:保留时间R
化合物2o:HPLC-MS:保留时间R
化合物2p:HPLC-MS:保留时间R
化合物2q:HPLC-MS:保留时间R
化合物2r:HPLC-MS:保留时间R
化合物2s:HPLC-MS:保留时间Rt=10.79min(梯度C);HRMS(ESI):m/z计算值:C
表2:通过碘环化的结合至固体载体的肽的氨基酸序列。在移除Fmoc基团、从树脂上裂解并通过制备型HPLC纯化后计算分离收率。该表显示经由鹅膏蕈碱的B环(N端Trp和C端Cys)进行环化的实施例(参见图2)。
分析数据:
化合物4a:HPLC-MS:保留时间R
化合物4b:HPLC-MS:保留时间R
化合物4c:HPLC-MS:保留时间R
化合物4d:HPLC-MS:保留时间R
化合物4e:HPLC-MS:保留时间R
化合物4f:HPLC-MS:保留时间R
化合物4g:HPLC-MS:保留时间R
化合物4h:HPLC-MS:保留时间R
化合物4i:HPLC-MS:保留时间R
化合物4j:HPLC-MS:保留时间R
化合物4k:HPLC-MS:保留时间R
化合物4l:HPLC-MS:保留时间R
化合物4m:HPLC-MS:保留时间R
化合物4n:HPLC-MS:保留时间R
化合物4o:HPLC-MS:保留时间R
化合物4p:HPLC-MS:保留时间R
化合物4q:HPLC-MS:保留时间R
化合物4r:HPLC-MS:保留时间R
化合物4s:HPLC-MS:保留时间R
化合物4t:HPLC-MS:保留时间R
化合物4u:HPLC-MS:保留时间R
化合物4v:HPLC-MS:保留时间R
表3.通过碘环化的结合至固体载体的肽的氨基酸序列。该表显示经由A环(N端Cys和C端Trp)进行环化的实施例(参见图3)。
分析数据:
化合物6a:HPLC-MS:保留时间R
化合物6b:HPLC-MS:保留时间R
化合物6c:HPLC-MS:保留时间R
化合物6d:HPLC-MS:保留时间R
化合物6e:HPLC-MS:保留时间R
化合物6f:HPLC-MS:保留时间R
化合物6g:HPLC-MS:保留时间R
化合物6h:HPLC-MS:保留时间R
表4.未通过碘环化的结合至固体载体的肽的氨基酸序列。
对于表4中包含的化合物,环化肽的收率低于5%。
表5:由单环前体合成的双环肽
#:Sar:肌氨酸;§:Aib:α-氨基异丁酸;(*):aze:氮杂环丁烷;(**)Tle:-叔-亮氨酸;(***):Acc:氨基环丙烷-1-羧酸
分析数据:
化合物7a:HPLC-MS:保留时间R
使2-CTC树脂(1g,0.98mmol/g)负载如所述的Fmoc-L-反式-Hyp(OTBS)-OH(参见材料和方法)。树脂载量测定为0.30mmol/g。根据方法A移除Fmoc基团。根据方法A和B,使以下肽序列Fmoc-L-Asn(Trt)-OH(4当量)、Fmoc-L-Cys(Trt)-OH(4当量)、Fmoc-Gly-OH(4当量)、Fmoc-L-Ile-OH(4当量)、Fmoc-Gly-OH(4当量)、Fmoc-L-Trp-OH(4当量)和Fmoc-L-(2S,3R,4R)-(TBS)
化合物7b:HPLC-MS:保留时间R
化合物7b的合成以与如上所公开的化合物7a类似的方式使用相应的Fmoc保护的氨基酸进行。使用制备型HPLC纯化粗产物,以获得呈白色粉末状的双环八肽7b(20mg,68%)。
化合物7c:HPLC-MS:保留时间R
化合物7c的合成以与化合物7a(Ama-03)的合成类似地使用相应的Fmoc保护的氨基酸进行。使用制备型HPLC纯化粗产物,以获得呈白色粉末状的双环八肽7c(30mg,60%)。
化合物7d:HPLC-MS:保留时间R
使2-CTC树脂(1g,0.98mmol/g)负载如所述的S24(参见材料和方法)。树脂载量测定为0.30mmol/g。根据方法A移除Fmoc基团。根据方法B,使Fmoc-Cys(Trt)-OH(4当量)与脱保护的树脂偶联。根据方法A移除所得的树脂的Fmoc基团。根据方法A和B,使以下肽序列Fmoc-Gly-OH(4当量)、Fmoc-L-Ile-OH(4当量)、Fmoc-Gly-OH(4当量)、Fmoc-L-Trp-OH(4当量)和Fmoc-L-(2S,3R,4R)-(TBS)
化合物7e:HPLC-MS:保留时间R
化合物7e的合成是7f(α-鹅膏蕈碱)的全合成的一部分并且在本文中公开。
化合物7f:HPLC-MS:保留时间R
a)DCC,NHS,DCM;b)H-Hyp-OH,NaHCO
使2-CTC树脂(1g,0.98mmol/g)负载如所述的S24(参见材料和方法)。树脂载量测定为0.30mmol/g。根据方法A移除Fmoc基团。根据方法B,使Fmoc-Cys(Trt)-OH(4当量)与脱保护的树脂偶联。根据方法A移除所得的树脂的Fmoc基团。根据方法A和B,使以下肽序列Fmoc-Gly-OH(4当量)、Fmoc-L-Ile-OH(4当量)、Fmoc-Gly-OH(4当量)、Fmoc-L-6-OBn-Trp-OH(4当量)和Fmoc-L-(2S,3R,4R)-(TBS)
HRMS(ESI):m/z C
HPLC-MS:保留时间R
将单环八肽S26(80mg,79μmol,1.0当量)溶解于DMF(40mL)中。然后,在0℃下添加DIPEA(30μL,2.2当量)和HATU(20mg,2.0当量)。使反应混合物温热至室温维持12小时并减压浓缩。使用制备型HPLC纯化粗产物,以获得呈白色粉末状的双环八肽S27(52mg,68%)。
HRMS(ESI):m/z计算值:C
HPLC-MS:保留时间R
将双环八肽S27(16mg,16μmol,1.0当量)溶解于EtSH(2mL)中,并在剧烈搅拌下用BF
HRMS(ESI):m/z计算值:C
HPLC-MS:保留时间R
将所获得的白色粉末6-OH-S-脱氧-鹅膏蕈碱(化合物7e)(8mg,8.8μmol,1.0当量)溶解于2mL的i-PrOH/EtOH混合物(2∶1)中。然后,滴加mCPBA(0.7当量)的i-PrOH/EtOH(2∶1)溶液。将所得的溶液搅拌30分钟,然后通过添加水(1mL)终止反应。经由制备型HPLC纯化粗产物,得到呈白色粉末状的α-鹅膏蕈碱(3mg,38%,化合物7f)。
HRMS(ESI):m/z计算值:C
HPLC-MS:保留时间R
化合物7g:HPLC-MS:保留时间R
化合物7g的合成以与如上所公开的化合物7a类似的方式使用相应的Fmoc保护的氨基酸进行,并用Fmoc-D-Pro-OH进行2-CTC-树脂的初始负载。使用制备型HPLC纯化粗产物,以获得呈白色粉末状的双环八肽7g(25mg,63%)。
化合物7h:HPLC-MS:保留时间R
化合物7h的合成以与如上所公开的化合物7a类似的方式使用相应的Fmoc保护的氨基酸进行。使用制备型HPLC纯化粗产物,以获得呈白色粉末状的双环八肽化合物7h(Ama-02)(22mg,64%)。
化合物7i:HPLC-MS:保留时间R
从肽基树脂S24开始,在位置4处使用Fmoc-D-Pro-OH代替Fmoc-Gly-OH来安置线性前体。根据在S29的合成中使用的相同操作,在RP-HPLC纯化之后获得呈白色固体粉末状的化合物7i(Ama-12)。
化合物7j:HPLC-MS:保留时间R
从肽基树脂S24开始,使用Fmoc-L-HomoCys(Trt)-OH代替Fmoc-L-Cys(Trt)-OH来安置线性前体。根据在S29的合成中使用的相同操作,在RP-HPLC纯化之后获得呈白色固体粉末状的化合物7i(Ama-14)。
化合物7k:HPLC-MS:保留时间R
a)DCC,NHS,DCM;b)H-Hyp-OH,NaHCO
化合物7l:保留时间R
从肽基树脂S24开始,在位置4处使用Fmoc-Sar-OH代替Fmoc-Gly-OH来安置线性前体。根据在S29的合成中使用的相同操作,在RP-HPLC纯化之后获得呈白色固体粉末状的化合物7l(Ama-13)。
化合物7m:保留时间R
从肽基树脂S24开始,在位置4处使用Fmoc-Aib-OH(N-α-Fmoc-α-氨基异丁酸)代替Fmoc-Gly-OH来安置线性前体。根据在S29的合成中使用的相同操作,在RP-HPLC纯化之后获得呈白色固体粉末状的化合物7m(Ama-15)。
化合物7n:HPLC-MS:保留时间R
化合物7n的合成:
使2-CTC树脂(1g,0.98mmol/g)负载如所述的Fmoc-L-Aze-OH(参见材料和方法)。树脂载量测定为0.30mmol/g。根据方法A移除Fmoc基团。根据方法B,使Fmoc-Asn(Trt)-OH(4当量)与脱保护的树脂偶联。进行氯醌测试以证实偶联完成。根据方法A移除所得的树脂的Fmoc基团。根据方法A和B,使以下肽序列Fmoc-L-Cys(Trt)-OH(4当量)、Fmoc-Gly-OH(4当量)、Fmoc-L-Ile-OH(4当量)、Fmoc-Gly-OH(4当量)和Fmoc-L-Trp-OH(4当量)与脱保护的树脂偶联。在固体载体上进行碘介导的环化(参见材料和方法)并使用条件A将肽从树脂上裂解之后,获得呈白色固体粉末状的37.2mg肽S11,在后续纯化后的收率为17%。向Fmoc-DHIle(TBS)
化合物7o:HPLC-MS:保留时间R
从肽基树脂S24开始,在位置4处使用Fmoc-D-Ala-OH代替Fmoc-Gly-OH来安置线性前体。根据在S29的合成中使用的相同操作,在RP-HPLC纯化之后获得呈白色固体粉末状的化合物7o(Ama-27)。
化合物7p:HPLC-MS:保留时间R
从肽基树脂S24开始,在位置3处使用Fmoc-L-Tle-OH代替Fmoc-L-Ile-OH来安置线性前体。根据在S29的合成中使用的相同操作,在RP-HPLC纯化之后获得呈白色固体粉末状的化合物7p(Ama-32)。
化合物7q:HPLC-MS:保留时间R
根据S11的合成,但使用Fmoc-L-4-Pro(F
向Fmoc-DHIle(TBS)
化合物7r:HPLC-MS:保留时间R
从肽基树脂S24开始,在位置4处使用Fmoc-Acc-OH代替Fmoc-Gly-OH来安置线性前体。根据在S29的合成中使用的相同操作,在RP-HPLC纯化之后获得呈白色固体粉末状的化合物7r(Ama-44)。
前体合成
(2S,4R)-1-((((9H-芴-9-基)甲氧基)羰基)-L-天冬酰胺酰基)-4-羟基吡咯烷-2-甲酸
在0℃下,将Fmoc-Asn(Trt)-OH(6g,10mmol)和N-羟基琥珀酰亚胺(NHS,1.15g,10mmol)添加到干燥DMF(25mL)中。在该温度下,缓慢添加DCC(2.16g,10.5mmol),由此形成白色沉淀。将反应混合物温热至室温,并搅拌12小时。将反应物倾注到冰水(20mL)上,用EtOAc萃取两次,并将有机萃取物用盐水洗涤,干燥(Na
HRMS(ESI):m/z计算值:C
HPLC-MS:保留时间R
S24:(2S,4R)-1-((((9H-芴-9-基)甲氧基)羰基)-L-天冬酰胺酰基)-4-((叔丁基二甲基甲硅烷基)氧基)吡咯烷-2-甲酸
在0℃下,将以上所获得的二肽(2g,4.2mmol)和DIPEA(1.5ml,8.5mmol)添加到干燥DMF(25mL)中。在该温度下,缓慢添加叔丁基二甲基甲硅烷基三氟甲磺酸酯(1.95mL,8.5mmol)。将反应混合物温热至室温,并搅拌12小时。将反应物倾注到冰水(20mL)上,用EtOAc萃取两次,并将有机萃取物用盐水洗涤,干燥(Na
HRMS(ESI):m/z计算值:C
HPLC-MS:保留时间R
Fmoc-L-Trp(6-OBn)-OH 15作为鹅膏蕈碱的关键构建单元以及有限的商业可得性,其合成是高度需要的。进行吲哚氨基甲酸酯的Vilsmeier-Haack甲酰化以形成醛,随后使其转化为Boc保护的吲哚9。随后进行Horner-Wadsworth烯化以形成脱氢氨基酸10。使用Rh(COD)
1)Horner-Wadsworth烯化
对于该步骤,通过使用Boc-2-膦酰甘氨酸-甲基二甲酯和DBU来合成(Z)-二脱氢氨基酸。存在一种替代方法,其使用四甲基胍(CAS 80-70-6)作为碱在THF中在-70℃至室温下进行。
2)氢化
对于该步骤,通过使用Rh(COD)
在0℃下,将三氯氧磷(1.5mL,15mmol)滴加到干燥DMF(5mL)中。在该温度下,缓慢添加含6-OBn吲哚(2.23g,10mmol)的干燥DMF(5mL),由此形成亮黄色沉淀。将反应混合物温热至45℃,并搅拌2小时。将反应物倾注到冰水(20mL)上,用乙醚萃取两次,并弃去醚萃取物。然后将水层用氢氧化钠水溶液处理直至溶液呈碱性,并用乙醚萃取。将有机萃取物用盐水洗涤,干燥(Na
向schmidt的膦酰甘氨酸酯(Boc-2-膦酰甘氨酸-甲基二甲酯)(3.04g,9.1mmol)的二氯甲烷(10mL)溶液中添加DBU(1.15mL,7.7mmol)。在搅拌10分钟后,缓慢添加含醛10(2.45g,7mmol)的二氯甲烷(10mL)。在将反应混合物搅拌16小时后,使溶剂在减压下蒸发。将残余物溶解于乙酸乙酯(40mL)中。然后,将有机溶液用1N HCl(2×10mL)和盐水洗涤,干燥(NaSO
在氩气气氛下,向11(1.04g,2mmol)的干燥脱气DCM(10mL)溶液中添加16mg(0.04mmol)[Rh(COD)
在0℃下,向甲酯12(1.07g,2.04mmol)的MeOH(4mL)和THF(4mL)溶液中添加0.5MLiOH水溶液(4mL)。使混合物温热至室温维持2小时,并在TLC显示原料完全消耗后真空移除溶剂。将残余物溶解于水中并用DCM洗涤。然后,将水层使用1M KHSO
为了测试不同的schmidt膦酰甘氨酸酯,也进行了Cbz保护的6-OBn-Trp的合成。
向schmidt的膦酰甘氨酸酯(Cbz-2-膦酰甘氨酸-甲基二甲酯)(3.01g,9.1mmol)的二氯甲烷(10mL)溶液中添加DBU(1.15mL,7.7mmol)。在搅拌10分钟后,缓慢添加含醛10(2.45g,7mmol)的二氯甲烷(10mL)。在将反应混合物搅拌16小时后,使溶剂在减压下蒸发。将残余物溶解于乙酸乙酯(40mL)中。然后,将有机溶液用1N HCl(2×10mL)和盐水洗涤,干燥(NaSO
在氩气气氛下,向16(1.11g,2mmol)的干燥脱气DCM(10mL)溶液中添加16mg(0.04mmol)[Rh(COD)
在氩气气氛下,向11(1.04g,2mmol)的干燥脱气DCM(10mL)溶液中添加16mg(0.04mmol)[Rh(COD)
材料和方法
HPLC-MS:
在与配备有C
溶剂梯度为梯度A、梯度B或梯度C:
梯度A:0-10分钟10%-50%B,10-13分钟100%B,13-16分钟20%B,
梯度B:0-10分钟20%-100%B,10-13分钟100%B,13-16分钟20%B。
梯度C:0-10分钟50%-100%B,10-13分钟100%B,13-16分钟20%B。
通过固相肽(SPPS)合成制备线性肽
树脂负载:使2-CTC树脂(1g,0.98mmol/g)在含有玻璃料的固相肽合成容器(10mL)的DCM中预溶胀20分钟。在排干溶剂后,将含Fmoc-氨基酸(根据氨基酸序列选择)Fmoc-Axx-OH或二肽Fmoc-Asn-Hyp(OTBS)-OH(0.3mmol)和DIPEA(4.5mmol)的DCM(3mL)添加到树脂中。将混合物搅拌2小时,然后排干溶剂。用DMF(4×3mL)洗涤树脂。然后,添加MeOH/DIPEA/DCM的混合物(1∶1∶8)以封闭与树脂结合的剩余2-氯三苯甲基氯。将混合物搅拌0.5小时。然后排干溶剂并用DMF(4×3mL)洗涤树脂。优选地,树脂用于进一步的肽合成,其中氨基酸的载量<0.30mmol/g。使用以下方案:
方法A)Fmoc基团的移除.将20%哌啶的DMF(3mL)溶液添加到树脂(1g;载量<0.30mmol/g)中,并将所得的悬浮液振荡10分钟。然后从树脂中移除溶液。再次,将20%哌啶的DMF(3mL)溶液添加到树脂中,并将所得的悬浮液再振荡10分钟。排干溶液并用DMF(6×3mL)洗涤树脂。
方法B)氨基酸偶联.将氨基酸(4.0当量)和TBTU(4.0当量)溶解于干燥DMF(3mL)中。将DIPEA(12当量)滴加到DMF溶液中。在活化1分钟后,将所得的溶液添加到Fmoc脱保护的树脂(1g;载量<0.30mmol/g)中。将混合物振荡直至偶联反应完成。然后,排干溶液并用DMF(4×3mL)冲洗树脂。
所有具有不同氨基酸序列的线性肽均使用该方案以Fmoc-脱保护(方法A)和氨基酸偶联(方法B)的交替顺序合成。
A)固相环化
硫醚桥(氨基羧乙基硫色氨酸基序)由线性树脂结合肽(1当量;(500mg;载量<0.30mmol/g))根据以上方法合成。在保护气氛(Ar或氮气)下,通过添加新鲜制备的碘的DMF(2当量,2mg/m1)溶液实现硫醚的形成。将混合物在氮气或氩气气氛下振荡2.5小时以完成硫醚的形成。如果需要,也应用更长的反应时间(测试裂解的HPLC-MS控制)。然后,排干溶液并用DMF(4×3mL)冲洗树脂。
B)溶液相环化
在氩气或氮气气氛下,向新鲜制备的碘的DMF(1当量,2mg/ml)溶液中添加线性肽(1当量)。然后,将溶液搅拌12小时以完成硫醚的形成(HPLC-MS控制!)。
从固体载体上裂解
条件A)将树脂(500mg;载量<0.30mmol/g)用10mL的TFA/TIS/H
条件B)将树脂(500mg;载量<0.30mmol/g)用10mL的TFE/HOAc/DCM的混合物(1∶1∶8)在室温和温和搅拌下处理2小时。将树脂过滤并用DCM(2×5mL)冲洗。合并冲洗液和滤液并蒸发至干。
条件C)(测试裂解):为了探索裂解反应的完成,从反应混合物中取出1-2mg树脂,将其提交至配备有玻璃料的塑料移液管。然后将树脂分别用1-2ml的DMF(2×)、DCM(2×)洗涤并干燥。将TFA(0.25mL)添加到树脂中并温育5分钟。然后真空移除TFA并将残余物用于HPLC-MS分析。
大内酰胺化
在0℃下,向DIPEA(5当量)的DMF溶液中的单环肽(1当量)溶液中添加HATU(2当量)。将溶液搅拌12小时,随后进行制备型HPLC纯化。将分离的产物冻干,以得到白色固体。
保护基的脱保护
去苄基化:将最终的苄基保护的鹅膏蕈碱或衍生物溶解于EtSH(10mg/ml)中并用N
脱甲硅基:方法A:向含TBDMS-保护的S-脱氧鹅膏蕈碱或衍生物的MeCN(10mg/ml)中添加BF
氧化
将含有氨基羧乙基硫色氨酸的肽溶解于iPrOH/EtOH 2∶1(50μL)中。在室温下,向所得的溶液中添加mCPBA(1.0当量)。通过HPLC-ESI-MS监测反应进程。在反应完成后(30分钟),将粗反应混合物用HPLC缓冲液稀释并在HPLC上直接纯化,以得到(R)-亚砜和(S)-亚砜的混合物(基于HPLC峰积分),其可通过制备型HPLC分离。
使用THP树脂通过I
使Fmoc-Hyp-OH(10.00g,28.3mmol)悬浮于100ml80%MeOH中并添加Cs
收率:9.85g,89%
将氨基酸19(9.85g,25.0mmol)、4-甲苯磺酸吡啶(2.62g,10.4mmol)添加到1,3-二氢-2H-吡喃-2-基-甲氧基甲基树脂(10.0g,0.77mmol/g THP树脂)在80ml二氯乙烷中的悬浮液中。将反应物在80℃下搅拌过夜。在冷却后,将树脂过滤,并用二氯乙烷、二甲基甲酰胺、乙腈、二氯甲烷和叔丁基甲基醚充分洗涤。
载量为0.60mmol/g(通过脱保护后的芴甲基的UV光谱来测定)。
树脂预处理:
将负载的树脂20(3.20g,1.92mmol)用N,N-二甲基巴比妥酸(5.51g,35.3mmol)和Pd(PPh
偶联操作:
将所有反应物和试剂溶解于二氯甲烷/DMF(1∶1,v/v)中,并使用CEM LibertyBlue肽合成仪以0.5mmol规模进行线性肽的合成。使用氨基酸/HOBt(各0.3N)、PyBOP(0.5N)和DIEA(2N)的储备溶液。使用2-5当量的氨基酸以1∶1∶1∶2的比率使用氨基酸∶HOBt∶PyBOP∶DIEA。肽偶联通过微波照射(50W,CEM微波反应器)在50℃下进行10分钟,并在偶联后用DMF洗涤。
Fmoc-脱保护:
通过在50℃下添加10.0ml20%哌啶的N,N-二甲基甲酰胺维持10分钟来进行脱保护。将树脂用DMF洗涤(在偶联最终氨基酸后未脱保护,因为其用作N
在完成后,将树脂最终转移到具有底部玻璃料的注射器中,用DCM洗涤并在减压下干燥。
碘介导的脱氧-鹅膏蕈碱前体的环化的一般操作
使THP树脂结合的线性前体肽21a(100μmol,1.0当量)在溶剂(15.6mL TFE/水9∶1或96mL DCM/TFA 95∶1)中溶胀,并在空气中5分钟的时段内将碘(100μmol,1.0当量)的DCM(4.0mL)溶液分批添加到所得的悬浮液中。将悬浮液在室温下搅拌2小时(溶剂=DCM/TFA)或19小时(溶剂=TFE/水)。添加乙二硫醇(85μL,1.0mmol,10当量,EDT)以淬灭反应,并将悬浮液过滤。将树脂用二氯甲烷(3×1分钟)洗涤,并将合并的有机层真空浓缩。将残余物在室温下用5.0mL DCM/TFA(1∶1)处理3小时。将淡褐色悬浮液滴入冰冷的甲基-叔丁基醚/己烷溶液(1∶1,45mL)中。将醚-肽-悬浮液在-20℃下温育30分钟,将悬浮液离心并将沉淀用冰冷的甲基-叔丁基醚/己烷(1∶1,80mL)洗涤一次。将分离的沉淀溶解在5.0mL的乙腈/水3∶1(+0.05%TFA)中,并通过离心移除不溶性颗粒。将沉淀如刚才所述再处理两次,并将合并的滤液冷冻干燥。通过制备型RP-HPLC纯化所得的粗制肽,获得呈无色TFA-盐的单环肽22a(溶剂TFE/水:34.7mg,28.9μmol,29%或溶剂DCM/TFA:31.6mg,26.3μmol,26%,基于初始树脂载量)。获得副产物(28.8-31.9mg),并表征为脱保护的二硫化物。RP-HPLC:t
使THP树脂结合的线性前体肽21b(250μmol,1.0当量)在二氯甲烷(240mLL)中溶胀,并小心地滴加三氟乙酸(TFA,2.5mL)。在空气中,在5分钟时段内,向所得的黄色悬浮液中分批添加碘(64.3mg,250μmol,1.0当量)的二氯甲烷(10mL)溶液。所得的肽浓度为1mM。将悬浮液在室温下搅拌2小时。随后,添加乙二硫醇(50μL,594μmol,2.3当量)和TIS(250μL)以淬灭反应。将悬浮液过滤,并将树脂用二氯甲烷(3×1分钟)洗涤。将合并的有机层真空浓缩,并将残余物在室温下用4.0mL的试剂R(TFA∶苯甲硫醚∶EDT∶苯甲醚90∶5∶3∶2)处理60分钟。将悬浮液滴入冰冷的甲基叔丁基醚/己烷溶液(1∶1,80mL)中,并在-20℃下温育30分钟。将悬浮液离心并将沉淀用冰冷的甲基-叔丁基醚/己烷(1∶1,80mL)洗涤一次。将分离的沉淀溶解在乙腈/水3∶1(+0.05%TFA)中,并通过离心移除不溶性颗粒。将沉淀如刚才所述再处理两次,并将合并的滤液冷冻干燥。通过制备型RP-HPLC纯化所得的粗制肽(262.4mg),获得呈无色TFA-盐的单环肽22b(95.1mg,79.1μmol,32%,基于初始树脂载量)。获得副产物(28.0mg,25.6μmol,10%),并表征为脱保护的线性肽,由裂解的二硫化物副产物生成。RP-HPLC:t
分析型RP-HPLC
柱:Luna 10μm C18(2),100A,250x4.6mm
梯度:溶剂A=水(+0,05%TFA);溶剂B=乙腈
0至25min:由5%至81%B;25至25.2min:由81%至100%B;25.2至27.5min:100%B;27.5至28min:由100%至5%B;28至30min:5%B
制备型RP-HPLC
柱:Luna 10μm C18(2),100A,250x21.2mm
梯度:溶剂A=水(+0,05%TFA);溶剂B=乙腈
0至20min:由5%至46%B;20至20.2min:由46%至100%B;20.2至22.5min:100%B;22.5至25min:5%B.
使用RNA聚合酶II抑制测定并通过在HEK细胞和HEK-OATP1B3细胞上的细胞生存力分析中评估细胞毒性,对本发明化合物的细胞毒性进行评估,其结果示出于表6中。
体外RNA Pol II测定使用
在人胚肾HEK293细胞和过表达有机阴离子转运多肽1B3(OATP1B3)的HEK293-OATP1B3细胞中,测定本发明化合物的体外毒性,以评估OATP1B3是否介导鹅膏蕈碱类似物向细胞中主动转运。将野生型HEK293(ATCC,Manassas,VA)和HEK293-OATP1B3细胞(参见:Pahl等人,Cytotoxic Payloads for Antibody-Drug Conjugates In:Amatoxins as RNAPolymerase II Inhibiting Antibody-Drug Conjugate(ADC)Payloads CHAPTER 19,TheRoyal Society of Chemistry 2019,ISBN:978-1-78801-077-1,或例如Bowman等人,DrugMetab Dispos 48:18-24,2020年1月)与化合物7f(α-鹅膏蕈碱)或如表6中所公开的化合物一起温育。细胞毒性通过BrdU分析进行测定。将HEK293和HEK-OATP1B3细胞以2.5×10
表6:细胞毒性
(*)表明在所测试的最高浓度下的残留RNAP II活性
文献
1.T.Wieland,Peptides of Poisonous Amanita Mushrooms(Eds.:A.Rich),Wiley-VCH,Weinheim,1986.
2.A.Pahl等人Drug Discov Today:Technol.2018,30,85-89.
3.M.O.Anderson,A.A.Shelat,R.K.Guy,J Org Chem 2005,70,4578-4584.
4.W.E.,Savige,A.Fontana,J.Chem.Soc.,Chem.Commun.1976,600-601.
5.G.Zanotti,L.Falcigno,M.Saviano,G.D′Auria,B.M.Bruno,T.Campanile,L.Paolillo,Chem-Eur J 2001,7,1479-1485.
6.K.Matinkhoo,A.Prvyma,M.Todorovic,B.O.Patrick,D.M.Perrin,J Am ChemSoc 2018,140,6513-651.
7.P.Sieber,B.Kamber,B.Riniker,W.Rittel,Helv Chim Acta 1980,63,2358-2363.
8.L.A.Schuresko,R.S.Lokey,Angew Chem Int Edit 2007,46,3547-3549.
9.G.Yao,J.O.Joswig,B.G.Keller,R.D.Süssmuth,Chem-Eur J 2019,25,8030-8034.
10.Edelman,G.M.等人,Proc.Natl.Acad.USA,63,78-85(1969)
11.Andreas Pahl,Christian Lutz,Torsten Hechler:Amatoxins as RNAPolymerase II Inhibiting Antibody-Drug Conjugate(ADC)Payloads,CHAPTER 19,TheRoyal Society of Chemistry 2019,ISBN:978-1-78801-077-1doi:10.1039/9781788012898-00398
12.Bowman等人Drug Metab Dispos 48:18-24,2020年1月
13.Binz等人Nat Biotechnol2005Oct;23(10):1257-68
14.Jain等人Pharm Res(2015)32:3526-3540
15.Dubowchik等人,Bioconjug Chem.13(2002)855-69
16.Peeters等人J Immunol Methods.1989Jun 2;120(1):133-43
17.Carlsson等人Biochem J.1978;173:723-37.