用于检测单细胞中全转录组的方法
文献发布时间:2023-06-19 19:23:34
技术领域
本公开是一种用于在单细胞水平上检测全转录组的新方法,其涉及全转录组的单细胞分析,并且特别涉及包括非编码RNA在内的单细胞全转录组的高通量检测。
背景技术
单细胞分析以单细胞分辨率测量DNA[1]、RNA[2]以及进行其他细胞分析。单细胞分析法能够有效地揭示样品内的异质性,并且产生更全面且准确的信息。单细胞分区、条形码化和高通量测序方面的最新技术进展使得并行检验来自数千个单细胞的基因的序列和表达谱变得可行[3]。这种高通量单细胞测序技术能够用于破译复杂的生物系统。目前,最常用的高通量单细胞测序方法是单细胞mRNA测序,其中通过测序定量检测各单细胞中mRNA的3′。然后可以使用单细胞中mRNA的表达谱来注释样品中的不同细胞类型,并且还可以用于发现各细胞中的基因和途径特征。通过单细胞mRNA测序产生的数据和见解(insights)极大地丰富了诸如癌症[4]、神经学[5]和免疫学[6]等不同领域的知识,并促进了疾病诊断和治疗的改进。
然而,大多数现有的单细胞mRNA测序方法依赖于通过RNA的3′poly-A尾与具有一段oligo-dT的寡核苷酸杂交来捕获RNA[7,8]。用这种方法无法检测没有poly-A尾的RNA种类。非编码RNA(ncRNA)为不编码蛋白质的一组转录物。长的非编码RNA(lncRNA)形成人转录组的大部分,并且在细胞和生理功能中发挥关键作用,例如染色质动力学、基因表达、细胞生长和分化[9]。肿瘤样品的全基因组相关研究(GWAS)已经证明大量lncRNA与多种癌症相关。lncRNA表达的变化及其突变促进肿瘤发生和转移,并且不同的lncRNA可能表现出肿瘤抑制和促进功能[10]。由于lncRNA的组织特异性表达特征和肿瘤学相关性,lncRNA可以用作癌症治疗的新的生物标志物和靶标。
微小RNA(miRNA)是大约20至22个核苷酸长的小的非编码RNA,其通过结合至mRNA的互补区域以抑制其翻译或调节其降解,从而在靶标基因的调节中发挥非常重要的作用[11]。这种调节似乎涉及许多基本的细胞过程,包括发育、分化、增殖、应激反应、代谢、凋亡和分泌[12]。其他ncRNA种类(例如snoRNA和环形RNA)都与细胞功能的各个方面有关。
非编码RNA表达分析的常规方法开始于从样品中提取总RNA,然后利用测序、微阵列或PCR分析总RNA或核糖体RNA删除的RNA[13,14]。大量样品中ncRNA的表达水平是样品中所有细胞类型的平均水平,其可以掩盖功能相关的细胞特异性ncRNA表达模式。尽管mRNA可以通过例如SMART-seq之类的方法在单细胞水平上进行常规检测,但是这些方法通常开始于用oligo-dT RT引物通过mRNA分子的3′poly-A尾捕获mRNA分子[15]。大多数ncRNA分子不具有poly-A尾,因此不能在单细胞水平上以这种方式被捕获。
一些现有方法可以捕获单细胞中的全转录组。然而,这些方法中的每一种都有其自身的缺点。
SUPeR-seq是这些方法中的一种。该方法用具有锚定序列的随机引物取代了常用的oligo-dT引物,并且可以同时捕获具有polyA尾和不具有polyA尾的RNA。该方法使用改进的细胞裂解和室温条件以避免捕获核糖体RNA(rRNA),rRNA可以占总RNA的约90%。由于在不同的细胞类型中,细胞组成可以是不同的,因此仍然需要测试该方法是否可以在不同细胞类型中有效地使rRNA捕获最小化[16]。
另一种方法RamDA-seq使用短的NSR(非随机引物)以捕获和逆转录RNA,同时排除rRNA。尽管该方法可以用于检测ncRNA,但是将NSR设计为短的寡核苷酸使得难以添加细胞条形码序列,从而使得该方法不适合用于在多个单细胞中并行检测ncRNA[17]。
发明内容
在本公开中,我们首先用一段具有特定序列(“标签”)的寡核苷酸延伸ncRNA的3′。可以通过酶或化学方法将标签添加到ncRNA的3′。可以以这样的方式设计RT引物,即RT引物可以通过添加的标签序列结合并捕获ncRNA。任选地,RT引物可以与能够作为细胞条形码的寡核苷酸序列结合,以将各单细胞与其他细胞区分开来,从而能够并行分析数千或更多单细胞。该方法也可以与微流体系统组合使用,其中可以将样品中的各细胞分割成单独的微室。单细胞可以在微室中裂解;然后可以添加标签序列,以便能够用标签特异性引物捕获ncRNA。
附图说明
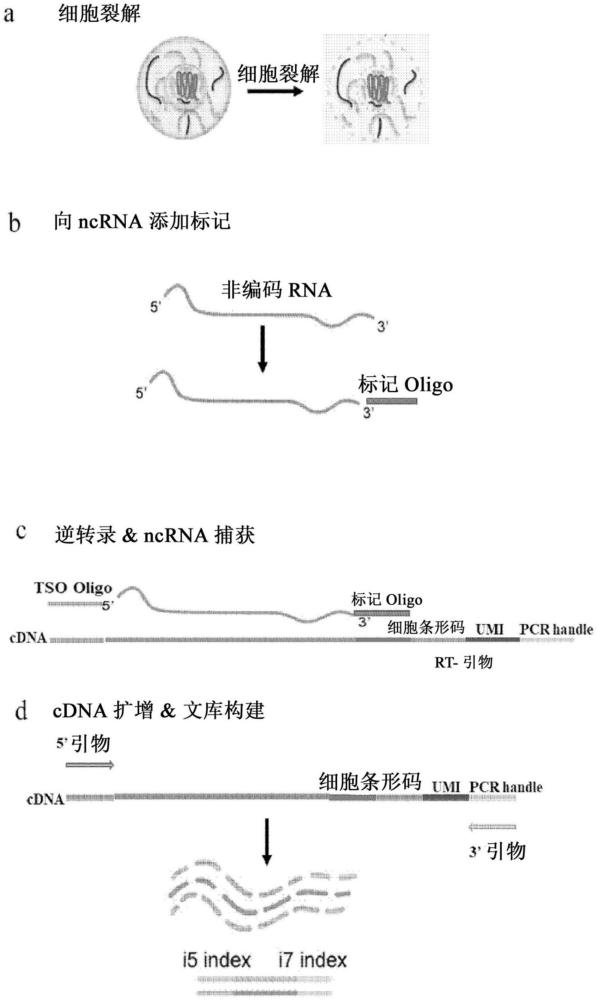
图1:本公开的示意图。
图2:本公开的实施方案的示意图,其中使用Poly(A)聚合酶向ncRNA添加polyA尾。
图3示出了UMI的百分比。
具体实施方式
为了克服现有的单细胞ncRNA分析方法的缺点,我们使用了ncRNA-方法以向ncRNA添加特定的标签序列。然后,该标签序列可以用于捕获ncRNA分子、和/或作为RT反应和扩增反应的引发位点(图1)。
本发明的一个实施方案是使用Poly(A)聚合酶向ncRNA添加polyA尾。然后,可以使用oligo-dT捕获和逆转录mRNA和具有新添加的polyA尾的ncRNA。如果在RT过程中引入模板转换寡核苷酸,则可通过PCR扩增所得的cDNA。利用独特的细胞条形码连同oligo-dT序列,可以标记来自同一单细胞的cDNA分子,并且可以并行处理一组单细胞,从而能够进行高通量单细胞分析(图2)。
将GEXSCOPE单细胞RNasq文库构建试剂盒(Singleron Biotechnologies)用于证明本公开在大规模并行单细胞ncRNA测序中的技术可行性和实用性。根据制造商的说明书进行实验,并进行如下改进。
简言之,将K562细胞的单细胞悬液加载到微芯片上,以将单细胞分配到芯片上的单个孔中。制备四个样品:两个用标准GEXSCOPE方案处理以用于单细胞mRNA测序(“对照”),两个用改进的方案进行以获得ncRNA读长(“nc”)。然后将细胞条形码化磁珠加载至微芯片并洗涤。每个细胞条码化磁珠的表面上包含具有独特的细胞条码序列的寡核苷酸,并结合了oligo-dT。珠粒上的每个寡核苷酸还具有独特的分子索引序列(UMI);在序列中检测到的UMI的数目可用于精确定量不同的RNA分子。根据珠粒和孔的直径(分别为约30μm和40μm),在微芯片上的每个孔中只能落入一个珠粒。使用下列反应混合物代替GEXSCOPE试剂盒中包含的裂解缓冲液,以裂解细胞并向ncRNA分子添加polyA尾。大肠杆菌(E.coli)Poly(A)聚合酶和10X大肠杆菌Poly(A)聚合酶反应缓冲液均来自New England Biolabs(NEB)。
将100μl反应混合物加载到芯片中,并在冰上孵育10分钟以裂解细胞。裂解细胞后,将微芯片在37℃孵育30分钟,以便向RNA的3'端添加PolyA尾。室温冷却30分钟后,将磁珠与捕获的RNA一起从微芯片取出,使用来自GEXSCOPE试剂盒的试剂,按照制造商的说明进行RT、模板转换、cDNA扩增和测序文库构建。在Illumina NovaSeq上用PE150模式对所得的单细胞RNAseq文库进行测序,并使用scopeTools生物信息学工作流程(SingleronBiotechnologies)进行分析。
如图3中所示,对应于总UMI中ncRNA的UMI百分比增加超过100%,平均值从1.5%变为3.6%。ncRNA UMI的显著增加的百分比证明了本公开的原理。此外,rRNA UMI的百分比保持相对低(0.9%,0.6%)。
参考文献
[1]Neu K E,Tang Q,Wilson P C,et al.Single-Cell Genomics:Approachesand Utility in Immunology[J].Trends in Immunology,2017,38(2):140-149.
[2]Byungjin H,Hyun L J,Duhee B.Single-cell RNA sequencingtechnologies and bioinformatics pipelines[J].Experimental&Molecular Medicine,2018,50(8):96.
[3]Klein A,Mazutis L,Akartuna I,et al.Droplet Barcoding for Single-Cell Transcriptomics Applied to Embryonic Stem Cells[J].Cell,2015,161(5):1187-1201.
[4]Baslan T,Hicks J.Unravelling biology and shifting paradigms incancer with single-cell sequencing[J].Nature reviews.Cancer,2017,17(9):557-569.
[5]Ofengeim D,Giagtzoglou N,Huh D,et al.Single-Cell RNA Sequencing:Unraveling the Brain One Cell at a Time[J].Trends in Molecular Medicine,2017,23(6).
[6]Papalexi E,Satija R.Single-cell RNA sequencing to explore immunecell heterogeneity[J].Nature Reviews Immunology,2017.
[7]Hashimshony,T.,Wagner,F.,Sher,N.&Yanai,I.CEL-Seq:single-cell RNA-Seq by multiplexed linear amplification.Cell Rep.2,666–673(2012).
[8]Ziegenhain C,Vieth B,Parekh S,et al.Comparative Analysis ofSingle-Cell RNA Sequencing Methods[J].Molecular Cell,2017,65(4):631-643.e4.
[9]Wu T,Du Y.LncRNAs:From Basic Research to Medical Application[J].International Journal of Biological Sciences,2017,13(3):295-307.
[10]Xiaoxia Ren.Genome-wide analysis reveals the emerging roles oflong non-coding RNAs in cancer.Oncol Lett.2020 Jan;19(1):588–594.
[11]Griffiths-Jones S,Grocock RJ,van Dongen S et al.miRBase:microRNAsequences,targets and gene nomenclature.Nucleic Acids Res 2006,34:D140-4.
[12]Wijnhoven BP,Michael MZ,Watson DI.MicroRNAs and cancer.Br J Surg2007,94(1):23-30.
[13]Nicole M White,Christopher R Cabanski,Jessica M Silva-Fisher,etal.Transcriptome sequencing reveals altered long intergenic non-coding RNAsin lung cancer[J].Genome Biology,15(8).
[14]Lopez J P,Diallo A,Cruceanu C,et al.Biomarker discovery:Quantification of microRNAs and other small non-coding RNAs using nextgeneration sequencing[J].Bmc Medical Genomics,2015,8(1):35.
[15]Picelli,Simone,Bjrklund,et al.Smart-seq2 for sensitive full-length transcriptome profiling in single cells[J].Nature Methods,2013,10(11):1096-1098.
[16]Fan,X.,Zhang,X.,Wu,X.et al.Single-cell RNA-seq transcriptomeanalysis of linear and circular RNAs in mouse preimplantation embryos.GenomeBiol 16,148(2015).https://doi.org/10.1186/s13059-015-0706-1