一种双途径合成壳寡糖的全细胞及其生产方法
文献发布时间:2023-06-19 19:28:50
技术领域
本发明涉及一种双途径合成壳寡糖的全细胞及其生产方法,属于合成生物学领域和生物医药领域。
背景技术
壳寡糖一般是指2~10个氨基葡萄糖以β-1,4糖苷键连接组成的相对分子质量壳聚糖。近年来,由于壳寡糖具有各种各样的生物活性,引起了人们的广泛关注。与壳聚糖相比,壳寡糖具有低粘度、高水溶性、生物相容性和可生物降解性等理化特征。到目前为止,壳寡糖在食品、人类健康、畜牧业和农业等研究领域得到了广泛的研究。特别是通过修饰得到的壳寡糖衍生物,由于其高效的传递效率,在药物开发领域具有巨大的潜力。
在过去的研究中,壳寡糖的制备主要通过壳聚糖降解获得,主要方法有化学法、酶解法和物理法等。化学法在酸解过程中,聚合度不容易控制且产量较低,还会造成严重的环境污染。采用物理法制备壳寡糖操作比较简单,但是效果比较差。另外,酶解法虽然反应条件比较温和,但是过程繁琐,成本较高,原材料成本较高。
研究发现,微生物细胞中存在以N-乙酰氨基葡萄糖(GlcNAc)和UDP-GlcNAc为前体,以几丁质合成酶NodC为催化剂的合成壳寡糖的生物合成途径。但是通常情况下,胞内UDP-GlcNAc和GlcNAc的浓度均不高,壳寡糖在单一细胞中的合成途径单一,无法满足工业化生产的需求。而且,目前尚未见基于多条途径合成壳寡糖的全细胞从头合成催化体系。
在前期研究过程中,本发明的发明人团队还开发了一种从头合成壳寡糖的多酶催化体系及其全细胞生产方法(CN114990174A),所述催化体系包含来源于Pseudomonasaeruginosa ATCC 15692的N-乙酰氨基葡萄糖激酶(AmgK)、来源于EscherichiacoliMG1655的UDP-N-乙酰葡糖胺焦磷酸化酶(GlmU)、来源于Azorhizobium caulinodansORS571的几丁质合成酶(NodC)和脱乙酰基酶(NodB)催化底物合成壳寡糖;该发明以大肠杆菌为底盘菌,通过自身相关基因的强启动子替换、重组质粒的快速构建与导入,获得拥有完整的壳寡糖从头合成途径的重组大肠杆菌,获得了具有从头合成壳寡糖途径的全细胞,可在优化后的催化体系中高效聚合葡萄糖或乙酰氨基葡萄糖为聚合度可控(DP<10)、分子量均一(~1000Da)的壳寡糖,在5L发酵罐体系中壳聚糖产量可达23.2g/L,在1000L发酵罐体系中壳聚糖产量达到20g/L以上。但是,该发明所采用的体外多酶催化体系生产成本很高,不利于工业生产;全细胞生产的壳寡糖也大多积聚在胞内,对于下游提取工艺提出了挑战。
目前,也有一些在枯草芽孢杆菌实现壳寡糖生产的报道。比如,有研究通过补偿途径的添加,在枯草芽孢杆菌中,壳寡糖的产量可达4.82g/L;但是该重组菌壳寡糖产量低,补偿途径冗杂(Ling,et al.2022,Metabolic Enginering)。
发明内容
为解决上述问题,本发明通过加强底物GlcNAc向细胞中的转运和缩短中间代谢路径,在枯草芽孢杆菌及酵母菌中构建新的代谢途径,实现了壳寡糖的胞外大量合成。本发明采用数据库挖掘的方式,获得了一系列酶元件,组装获得一条完整的微生物胞内合成壳寡糖的新途径。实现了该途径在工程菌株中的合理组装,从而获得了以葡萄糖和N-乙酰氨基葡萄糖(GlcNAc)为前体的双途径合成壳寡糖的全细胞转化方法。本发明的目的在于提供一种壳寡糖合成的新途径设计、全细胞转化的基因工程菌构建方法及壳寡糖的全细胞转化方法。
本发明的第一个目的是提供一种基因工程菌,所述基因工程菌过表达了5种酶:N-乙酰氨基葡萄糖转运蛋白(NagP)、N-乙酰氨基葡萄糖激酶(NagK)、6-P-N-乙酰氨基葡萄糖变位酶(PGM)、几丁质合成酶(NodC)、脱乙酰基酶(NodB)。
在一种实施方式中,所述NagP的氨基酸序列是NCBI上CAP50125所示的序列;所述NagK的氨基酸序列是NCBI上CCI76387所示的序列;PGM的氨基酸序列是NCBI上BAB00614所示的序列;NodC的氨基酸序列是NCBI上BAF89814所示的序列;NodB的氨基酸序列是NCBI上XP_003323413所示的序列。
在一种实施方式中,所述NagP的基因序列,优化密码子后,如SEQ ID No.1所示;所述NagK的基因序列,优化密码子后,如SEQ ID No.2所示;所述PGM的基因序列,优化密码子后,如SEQ ID No.3所示;所述NodC的基因序列,优化密码子后,如SEQ ID No.4所示;所述NodB的基因序列,优化密码子后,如SEQ ID No.5所示。该优化后的序列,适用于在大肠杆菌及枯草芽孢杆菌宿主中进行表达。
在一种实施方式中,所述NagP的基因序列,优化密码子后,如SEQ ID No.8所示;所述NagK的基因序列,优化密码子后,如SEQ ID No.9所示;所述PGM的基因序列,优化密码子后,如SEQ ID No.10所示;所述NodC的基因序列,优化密码子后,如SEQ ID No.11所示;所述NodB的基因序列,优化密码子后,如SEQ ID No.12所示。该优化后的序列,适用于在酿酒酵母宿主中进行表达。
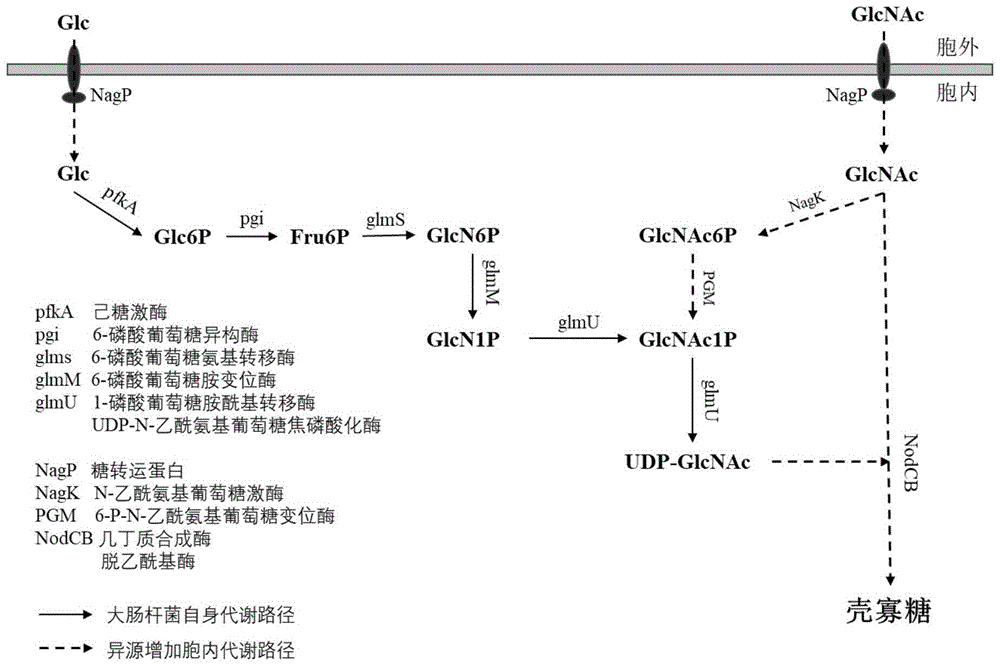
本发明的基因工程菌中,含有以葡萄糖为前体合成壳寡糖的多酶催化途径1,主要包含己糖激酶(pfkA)、6-磷酸葡萄糖异构酶(pgi)、6-磷酸葡萄糖氨基转移酶(glmS)、6-磷酸葡萄糖胺变位酶(glmM)、1-磷酸葡萄糖胺酰基转移酶(glmU)、UDP-N-乙酰氨基葡萄糖焦磷酸化酶(glmU)、几丁质合成酶(NodC)、脱乙酰基酶(NodB)七种酶蛋白。其中,pfkA、pgi、glmS、glmM、glmU、glmU对应的途径是微生物(大肠杆菌、枯草芽孢杆菌、酿酒酵母)自身存在的酶催化反应路径。
本发明的基因工程菌中,还含有以N-乙酰氨基葡萄糖为前体合成壳寡糖的多酶催化途径2,该途径是设计组合形成微生物(大肠杆菌、枯草芽孢杆菌、酿酒酵母)胞内壳寡糖合成的新途径;主要包含N-乙酰氨基葡萄糖转运蛋白(NagP)、N-乙酰氨基葡萄糖激酶(NagK)、磷酸化N-乙酰氨基葡萄糖变位酶(PGM)、UDP-N-乙酰氨基葡萄糖焦磷酸化酶(glmU)、几丁质合成酶(NodC)、脱乙酰基酶(NodB)六种酶蛋白。
在一种实施方式中,所述基因工程菌为大肠杆菌基因工程菌、枯草芽孢杆菌基因工程菌和酿酒酵母基因工程菌。
本发明的第二个目的是提供所述基因工程菌的构建方法。
在一种实施方式中,所述基因工程菌为大肠杆菌基因工程菌,构建方法如下:
(1)将NagP基因整合到pACtrc载体中,获得无诱导的表达重组质粒pACtrc-NagP;
(2)将NagK和PGM两种基因整合到pTrc100质粒中,获得无诱导的表达重组质粒pTrc100-NagK-PGM;
(3)将NodCB基因与载体pBBR-J23119质粒中,获得携带强启动子的无诱导表达质粒pBBR-J23119-NodCB;
(4)将上述三种重组质粒转入大肠杆菌细胞中,获得含有壳寡糖合成新途径的大肠杆菌基因工程菌。
在一种实施方式中,所述基因工程菌为枯草芽孢杆菌基因工程菌,构建方法如下:
(1)以RBS(CTTTAAGAAGGAGATATACAT,SEQ ID No.46)为NodCB基因的共表达核糖体结合位点,设计引物获得Gibson组装片段A;
(2)设计NagP基因的引物,并将其克隆到商业化载体pBE-S(Takara)多克隆位点MluI和BamHI中,构建获得重组质粒pBE-S-NagP;重新设计引物,以该重组质粒为模板,PCR获得Gibson组装的片段B;
(3)以RBS序列(TGCAAAGGAAAACAAACGCT,SEQ ID No.17)为NagK和PGM基因的共表达核糖体结合位点,设计引物获得Gibson组装片段C;
(4)以表达载体pP43NMK为模板,设计引物,以反向PCR的方法获得表达载体片段D,以上述获得的四个片段A、B、C、D通过体外重组的方式,并转化大肠杆菌,平板筛选获得最终表达质粒pP43-NodCB-NagP-NagK-PGM;通过电转化的方法转入表达宿主Bacillussubtilis 168菌株中。
在一种实施方式中,所述基因工程菌为酿酒酵母基因工程菌,构建方法如下:
(1)首先将密码子优化后的NagP基因克隆到质粒pGAPZA(Invitrogen)的多克隆位点EcoR I和Sal I处,构建获得重组质粒pGAPZA-NagP;以该质粒为模板,重新设计引物,将PCR产物克隆到酿酒酵母细胞的表达载体pRS426多克隆位点KpnI和XhoI中,构建获得表达质粒pRS426-NagP;
(2)以(GGGGS)2序列(如SEQ ID No.47)作为NagK和PGM基因的融合表达柔性linker,全基因合成并密码子优化;将该序列同样克隆到质粒pGAPZA(Invitrogen)的多克隆位点EcoR I和Sal I处,构建获得重组质粒pGAPZA-NagK-PGM;以该重组质粒为模板,重新设计引物,将PCR产物克隆到酿酒酵母细胞的表达载体pRS426--NagP多克隆位点XhoI和HindIII中,构建获得表达质粒pRS426-NagP-NagK-PGM;
(3)同理,以(GGGGS)2(如SEQ ID No.47)序列作为NodC和NodB基因的融合表达柔性linker,全基因合成并密码子优化。将该序列同样克隆到质粒pGAPZA(Invitrogen)的多克隆位点EcoRI和SalI处,构建获得重组质粒pGAPZA-NodCB;以该重组质粒为模板,重新设计引物,将PCR产物克隆到酿酒酵母细胞的表达载体pRS426--NagP-NagK-PGM多克隆位点HindIII和NotI中,构建获得表达质粒pRS426-NagP-NagK-PGM-NodCB;
(4)将上述构建成功的重组表达质粒pRS426-NagP-NagK-PGM-NodCB电转化到宿主酿酒酵母细胞BY4741,并在尿嘧啶缺陷型平板中筛选获得阳性克隆子。
本发明的第三个目的是提供壳寡糖合成的全细胞生产方法,所述方法是利用本发明的基因工程菌。
在一种实施方式中,所述方法是将上述的基因工程菌接种至液体种子培养基,最后逐级放大培养至高密度(OD
在一种实施方式中,所述的抗性平板配方为:15g/L琼脂粉,10g/L蛋白胨,5g/L酵母膏,10g/LNaCl,25mg/L四环素,80mg/L氨苄青霉素,25mg/L卡纳霉素。
在一种实施方式中,所述的大肠杆菌基因工程菌全细胞培养基配方为:10g/L蛋白胨,5g/L酵母膏,10g/LNaCl,10-100g/L葡萄糖或甘油,25mg/L四环素,80mg/L氨苄青霉素,25mg/L卡纳霉素)。维生素混合液(每升含有2mgbiotin,2mg folic acid,10mgpyridoxine·HCl,5mg riboflavin,5mg nicotinic acid,5mg calcium D-(+)-pantothenate,0.1mg vitamin B12,5mg p-aminobenzoic acid,5mg thioctic acid),10mL微量元素混合液(每升含有0.5g MnSO4·H2O,0.2g ZnSO4,0.1g CaCl2,0.1g CoCl2·6H2O,0.1g FeSO4·7H2O,20mg CuCl2·2H2O,10mg Na2SeO3,10mgNiCl2·6H2O,10mgNa2WO4·2H2O),采用HCl或NaOH调节最终pH7.2。
在一种实施方式中,所述的全细胞催化转化体系为50-80OD600,10-100g/L葡萄糖和乙酰氨基葡萄糖匀速流加。
在一种实施方式中,所述全细胞生产方法使用的是本发明的枯草芽孢杆菌基因工程菌或者酿酒酵母基因工程菌。
在一种实施方式中,所述全细胞生产方法具体是:采用枯草芽孢杆菌基因工程菌,活化后转接入发酵罐中,进行补料培养;其中发酵培养基由20g/L葡萄糖、20g/L酵母提取物、6g/L(NH
在一种实施方式中,所述全细胞生产方法具体是:采用酿酒酵母基因工程菌,活化后转接入发酵罐中,进行补料培养;其中,发酵培养基由60g/L葡萄糖、40g/L酵母提取物、微量矿物质和维生素构成;微量矿物质(g/L):(NH
本发明的第四个目的是提供以葡萄糖为前体合成壳寡糖的多酶催化途径,主要包含己糖激酶(pfkA)、6-磷酸葡萄糖异构酶(pgi)、6-磷酸葡萄糖氨基转移酶(glmS)、6-磷酸葡萄糖胺变位酶(glmM)、1-磷酸葡萄糖胺酰基转移酶(glmU)、UDP-N-乙酰氨基葡萄糖焦磷酸化酶(glmU)、几丁质合成酶(NodC)、脱乙酰基酶(NodB)七种酶蛋白。
本发明的第五个目的是以N-乙酰氨基葡萄糖为前体合成壳寡糖的多酶催化途径,该途径是设计组合形成微生物(大肠杆菌、枯草芽孢杆菌、酿酒酵母)胞内壳寡糖合成的新途径;主要包含N-乙酰氨基葡萄糖转运蛋白(NagP)、N-乙酰氨基葡萄糖激酶(NagK)、磷酸化N-乙酰氨基葡萄糖变位酶(PGM)、UDP-N-乙酰氨基葡萄糖焦磷酸化酶(glmU)、几丁质合成酶(NodC)、脱乙酰基酶(NodB)六种酶蛋白。
本发明还提供所述基因重组工程菌或多酶催化途径在生物化工、制药或食品领域中实现壳寡糖的高效合成的应用。
本发明的有益效果:
本发明以大肠杆菌、枯草芽孢杆菌、酿酒酵母为例,做为底盘菌,通过构建重组质粒导入宿主细胞中,设计组装了以N-乙酰氨基葡萄糖为前体合成壳寡糖的代谢路径二。同时利用微生物胞内自身存在的部分代谢路径一,构建了协同合成壳寡糖的全细胞(图1)。该重组细胞能够以葡萄糖和N-乙酰氨基葡萄糖为前体合成聚合度可控(DP<10)、分子量均一(~1000Da)的壳寡糖,展现了巨大的商业应用潜力。本发明采用全细胞生产的壳寡糖,最终获得胞外壳寡糖的含量可达20g/L,纯度较高,对于下游提取工艺降低了成本。
附图说明
图1是以葡萄糖和N-乙酰氨基葡萄糖合成壳寡糖的全细胞(大肠杆菌、枯草芽孢杆菌、酿酒酵母)代谢途径。
图2是5L发酵罐以五单位壳寡糖合成为代表的产物HPLC图谱特征峰。
图3是5L发酵罐以葡萄糖和N-乙酰氨基葡萄糖为前提的大肠杆菌基因工程菌合成壳寡糖的产量结果。
图4是5L发酵罐以葡萄糖和N-乙酰氨基葡萄糖为底物的枯草芽孢杆菌基因工程菌胞外合成壳寡糖的产量结果。
图5是5L发酵罐以葡萄糖和N-乙酰氨基葡萄糖为底物的酿酒酵母基因工程菌胞外合成壳寡糖的产量结果。
具体实施方式
下面结合附图和具体实施例对本发明作进一步说明,以使本领域的技术人员可以更好地理解本发明并能予以实施,但所举实施例不作为对本发明的限定。
下述实施例中,没有多作说明的都是采用常规分子生物学实验方法完成,所采用酶相关试剂盒均在大连宝生物公司购买。实施例中所涉及PCR、酶切、连接过程都是本领域技术人员根据产品说明书或本领域基础知识可以理解并且容易实现的,因此不再详细描述。
下述实施例中,所表达的NagP的氨基酸序列是NCBI上CAP50125所示的序列;NagK的氨基酸序列是NCBI上CCI76387所示的序列;PGM的氨基酸序列是NCBI上BAB00614所示的序列;NodC的氨基酸序列是NCBI上BAF89814所示的序列;NodB的氨基酸序列是NCBI上XP_003323413所示的序列。用于大肠杆菌及枯草芽孢杆菌宿主中进行表达时,进行了密码子优化;其中,所述NagP的基因序列如SEQ ID No.1所示;NagK的基因序列如SEQ ID No.2所示;PGM的基因序列如SEQ ID No.3所示;NodC的基因序列如SEQ ID No.4所示;NodB的基因序列如SEQ ID No.5所示。用于酿酒酵母宿主中进行表达时,进行了密码子优化;其中,所述NagP的基因序列如SEQ ID No.8所示;NagK的基因序列如SEQ ID No.9所示;PGM的基因序列如SEQ ID No.10所示;NodC的基因序列如SEQ ID No.11所示;NodB的基因序列如SEQ IDNo.12所示。
以大肠杆菌基因工程菌为例,以下实施例中以大肠杆菌JM109为质粒克隆扩增的主要载体,以大肠杆菌BL21(DE3)为酶表达宿主。所采用的质粒来源为商业化载体pBBR1MCS-2、pACYC184和pTrc99A的衍生质粒pActrc(pActrc是在pACYC184中lacI缺失,无需诱导的高强度表达质粒)、pTrc100(pTrc100是在pTrc99A中添加了trc启动子及rrnB T1终止子,无需诱导的高强度表达质粒)和pBBR-J23119(pBBR-J23119是在pBBR1MCS-2中替换了强启动子J23119,无需诱导);其中,pActrc、pTrc100、pBBR-J23119的具体的构建方法已经在CN114990174A中公开了。所采用质粒提取试剂盒为上海生工公司商业化产品;基因与引物的合成都由上海生工公司完成;大肠杆菌生物量即为细胞浓度,采用分光光度计于1cm光程测定600nm处的吸光度差值。
实施例1:具有壳寡糖合成途径的大肠杆菌基因工程菌全细胞构建方法
为实现壳寡糖从头合成途径多个关键酶在无外源诱导条件下的高效表达,需要联合三种兼容性质粒(pTrc100、pActrc、pBBR-J23119)共同转化大肠杆菌底盘细胞,同时采用高强度杂合启动子trc和J23119作为主要启动子。
优先以Azorhizobium caulinodans来源的NodC和Puccinia graminis来源的NodB为表达对象,进行全基因合成并进行密码子优化,以RBS(CTTTAAGAAGGAGATATACAT,如SEQID No.46)为NodCB基因的共表达核糖体结合位点,NodCB全基因序列如SEQ ID NO.6。设计克隆的引物,(nodCBF:5’-ATCC
以Xanthomonas campestris pv.campestris来源的N-乙酰氨基葡萄糖转运蛋白NagP为表达对象,进行全基因合成并进行密码子优化,设计NagP基因的引物(NagP,上游5’-ATCG
以RBS序列(TGCAAAGGAAAACAAACGCT,SEQ ID No.17)为NagK和PGM基因的共表达核糖体结合位点,全基因合成并密码子优化后全基因序列如SEQ ID No.7,设计NagK-RBS-PGM基因的引物(上游5’-ATCG
提取三种质粒(pBBR-J23119-NodCB、pACtrc-NagP、pTrc100-NagK-PGM),采用1-10ng的浓度均匀混合到100μl电击转化感受态细胞E.coli BL21(DE3),将含有大肠杆菌和质粒的预冷电击杯(间距1cm)擦干,迅速放入Bio-Rad公司生产的高压电击仪的电击槽;电击完成后,迅速取出电击杯并加入1mL无抗生素LB液体培养基,并全部转移到2mL无菌试管中;在30℃和150rpm摇床培养1小时后,4000rpm离心30s收集菌泥,全部涂布抗性平板(10g/L蛋白胨,5g/L酵母提取物,10g/LNaCl,15g/L琼脂粉,25mg/L四环素,100mg/L氨苄青霉素,25mg/L卡纳霉素),30℃培养18-22小时后筛选阳性单克隆,即具有壳寡糖合成途径二的重组大肠杆菌。同时该大肠杆菌中包含了壳寡糖合成途径一。
实施例2:大肠杆菌基因工程菌的培养条件及壳寡糖合成转化优化
根据实施例1的设计原理,采用两种代谢途径,以葡萄糖和N-乙酰氨基葡萄糖为前体底物,通过上述全细胞的发酵及转化,高效累积壳寡糖。
首先,在三抗性LB平板(15g/L琼脂粉,10g/L蛋白胨,5g/L酵母膏,10g/LNaCl,25mg/L四环素,100mg/L氨苄青霉素,25mg/L卡纳霉素)上挑取重组菌单克隆,接种至含有5mL种子培养基的试管中,在30℃和200rpm恒温摇床中培养8小时,获得重组大肠杆菌初步活化的种子液。发酵温度为30℃,搅拌转速为100-600rpm,通过流加策略(恒速流加和指数流加)直接将前体物质葡萄糖及N-乙酰氨基葡萄糖(比例优化1:3,1:2,1:1)加入到发酵液中,获得50L的高密度发酵产物。实现壳寡糖的大量积累,胞内产量达8g/L以上。同时,以在原始菌株宿主菌大肠杆菌BL21(DE3)为对照,采用相同的方法进行发酵,结果显示,因缺乏相应的外源基因,如NodCB,宿主菌大肠杆菌BL21(DE3)无法生产壳寡糖。
实施例3:双途径合成壳寡糖的枯草芽孢杆菌基因工程菌的构建及其在生产壳寡糖的应用
1、枯草芽孢杆菌基因工程菌的构建
为实现壳寡糖在枯草芽孢杆菌中的全合成,本发明以三种不同的组成型启动子体外分别构建三个表达盒,通过Gibson组装的方式获得重组表达质粒,从而转入枯草芽孢杆菌Bacillus subtilis 168菌株中进行表达。
设计NagP基因的引物,NagPF:5’-ACGCGTATGACTACAGCAAGGCCAGC-3’(SEQIDNo.20)和NagPR:5’-CGCGGATCCTTATTTCGGTTGTGCAGGCAG-3’(SEQ ID No.21),并将其克隆到商业化载体pBE-S(Takara)多克隆位点MluI和BamHI中,经过平板筛选,构建获得重组质粒pBE-S-NagP。重新设计引物,以该重组质粒为模板,上游:5’-ACTAGTGTTCTTTTCTGTATG-3’(SEQ ID No.22),下游:5’-AGTGTGTTATACTTTACTTGGAAG-3’(SEQ ID No.23)。PCR获得Gibson组装的片段B。
以RBS序列(TGCAAAGGAAAACAAACGCT,SEQ ID No.17)为NagK和PGM基因的共表达核糖体结合位点,全基因合成并密码子优化后全基因序列如SEQ ID No.7,设计NagK-RBS-PGM基因的引物(上游5’-ATCGCATATGCTATGTACTATGGTTTTGATATAGGG-3'(SEQ ID No.24),下游5’-CGCGGATCCTTACTTTACCAGCTCCGAC-3'(SEQ ID No.25)PCR扩增得到DNA片段并回收。采用两种限制性内切酶NdeI和BamHI分别对上述NagK-RBS-PGM基因片段和提取好的环状pMA5质粒(湖南丰晖生物科技有限公司)进行双酶切,通过琼脂糖凝胶电泳回收具有相同粘性末端的NagK-RBS-PGM基因和线性化质粒pMA5。酶切产物回收后进行T4 DNAligase酶连接和化学感受态细胞转化,氨苄青霉素抗性平板(10g/L蛋白胨,5g/L酵母提取物,10g/LNaCl,15g/L琼脂粉,100mg/L氨苄青霉素)筛选得到含有正确质粒pMA5-NagK-PGM的单克隆。重新设计引物,以该重组质粒为模板,上游:5’-CTTCCAAGTAAAGTATAACACACTtactacctgtcccttgctg-3’(SEQ ID No.26),下游:5’-gctatgaccatgattacgccaagcttcgcgcgaggcagagcttggaattg-3’(SEQ ID No.27)。PCR获得Gibson组装的片段C。
以RBS(CTTTAAGAAGGAGATATACAT,如SEQ ID No.46)为NodCB基因的共表达核糖体结合位点,NodCB全基因序列如SEQ ID NO.6。设计克隆的引物,(nodCBF:5’-ttataggtaagagaggaatgtacacATGAGCGTTGTTGATGTGATTGG-3'(SEQ ID No.28),nodCBR:5’-TTTCATACAGAAAAGAACACTAGTTTAATTATCGGTAGGCGCTGG-3'(SEQ ID No.29),PCR扩增得到Gibson组装的片段A。
以表达载体pP43NMK(湖南丰晖生物科技有限公司)为模板,设计引物,以反向PCR的方法获得表达载体片段D,上游:5’-aagcttggcgtaatcatggtcatagc-3’(SEQ ID No.30),下游:5’-gtgtacattcctctcttacctataa-3’(SEQ ID No.31)。
以上述获得的四个片段A、B、C、D通过体外重组的方式,并转化大肠杆菌,平板筛选获得最终表达质粒pP43-NodCB-NagP-NagK-PGM。通过电转化的方法转入表达宿主Bacillussubtilis 168菌株中,经筛选验证获得转化成功的枯草芽孢杆菌基因工程菌。
2、利用枯草芽孢杆菌基因工程菌生产壳寡糖
从琼脂平板上挑取单菌株接种在10mL LB培养基中培养,在37℃条件下以180rpm的转速振荡12小时。将培养物以3%接种量接种到含有100mL LBG培养基的500ml摇瓶中,然后在37℃下以200rpm条件下进行培养。16小时后,将种子培养物接种到5L发酵罐中,该发酵罐含有1900mL发酵培养基,发酵培养基由20g/L葡萄糖、20g/L酵母提取物、6g/L(NH
最终在枯草芽孢杆菌基因工程菌中,5L发酵罐体系下,48h胞外壳寡糖的含量达12g/L。
实施例4:双途径合成壳寡糖的酿酒酵母基因工程菌的构建及其在生产壳寡糖的应用
1、酿酒酵母基因工程菌的构建
为实现壳寡糖在酵母细胞中的从头合成,本发明采用组成型表达启动子pGAP在酿酒酵母细胞BY4741(北京华越洋生物)中进行多基因的克隆及表达。
首先将密码子优化后的NagP基因克隆到质粒pGAPZA(Invitrogen)的多克隆位点EcoR I和Sal I处,所采用的引物序列为NagPF:5’-CCGGAATTCATGACCACTGCTAGACCAGCC-3’(SEQ ID No.32)和NagPR:5’-ACGCGTCGACTCACTTTGGTTGAGCCGGCAAACC-3’(SEQ ID No.33)。构建获得重组质粒pGAPZA-NagP。以该质粒为模板,重新设计引物,SCNagPF:5’-CGGGGTACCTTTTTGTAGAAATGTCTTGGTGTCC-3’(SEQ ID No.34)和SCNagPR:5’-CCGCTCGAGTCTCACTTAATCTTCTGTACTCTGAAG-3’(SEQ ID No.35),将PCR产物克隆到酿酒酵母细胞的表达载体pRS426多克隆位点KpnI和XhoI中,构建获得表达质粒pRS426-NagP。
以(GGGGS)2序列(如SEQ ID No.47)作为NagK和PGM基因的融合表达柔性linker,全基因合成并密码子优化后全基因序列如SEQ ID No.36。将该序列同样克隆到质粒pGAPZA(Invitrogen)的多克隆位点EcoR I和Sal I处,所采用的引物序列为NagK-PGMF:5’-CCGGAATTCATGTACTACGGTTTCGATATTGG(SEQ ID No.37)和NagK-PGMR:5’-ACGCGTCGACTCACTTGACCAATTCCGAGACGG(SEQ ID No.38)。构建获得重组质粒pGAPZA-NagK-PGM。以该重组质粒为模板,重新设计引物SCNagK-PGMF:5’-CCGCTCGAGTTTTTGTAGAAATGTCTTGGTGTCC(SEQ IDNo.39)和SCNagK-PGMR:5’-CCCAAGCTTTCTCACTTAATCTTCTGTACTCTGAAG(SEQ ID No.40),将PCR产物克隆到酿酒酵母细胞的表达载体pRS426--NagP多克隆位点XhoI和HindIII中,构建获得表达质粒pRS426-NagP-NagK-PGM。
同理,以(GGGGS)2序列(如SEQ ID No.47)作为NodC和NodB基因的融合表达柔性linker,全基因合成并密码子优化后全基因序列如SEQ ID No.41。将该序列同样克隆到质粒pGAPZA(Invitrogen)的多克隆位点EcoRI和SalI处,所采用的引物序列为NodCBF:5’-CCGGAATTCATGTCTGTTGTCGATGTC(SEQ ID No.42)和NodCBR:5’-ACGCGTCGACTCAGTTGTCAGTTGGAGCTGG(SEQ ID No.43)。构建获得重组质粒pGAPZA-NodCB。以该重组质粒为模板,重新设计引物SCNodCBF:5’-CCCAAGCTTTTTTTGTAGAAATGTCTTGGTGTCC(SEQ ID No.44)和SCNodCBR:5’-ATTTGCGGCCGCTCTCACTTAATCTTCTGTACTCTGAAG(SEQ IDNo.45),将PCR产物克隆到酿酒酵母细胞的表达载体pRS426--NagP-NagK-PGM多克隆位点HindIII和NotI中,构建获得表达质粒pRS426-NagP-NagK-PGM-NodCB。
将上述构建成功的重组表达质粒pRS426-NagP-NagK-PGM-NodCB电转化到宿主酿酒酵母细胞BY4741,并在尿嘧啶缺陷型平板中筛选获得阳性克隆子。筛选平板培养基(1L)包括22g葡萄糖,6.7gYNB(无氨基酸酵母氮源),Leu(亮氨酸)0.1g,Trp(色氨酸)0.02g,His(组氨酸)0.02g,加入1M NaOH溶液调节pH至6.0左右。若配制固体培养基,加入20g琼脂粉,115℃,灭菌15min。
2、利用酿酒酵母基因工程菌生产壳寡糖
将上述得到的阳性克隆子接种到100ml种子培养基(YPD)中,30℃培养18h后,接种到5L发酵罐培养,该发酵罐含有1900mL发酵培养基,发酵培养基由60g/L葡萄糖、40g/L酵母提取物、微量矿物质和维生素构成。微量矿物质(g/L):(NH
最终在酿酒酵母基因工程菌中,5L发酵罐体系下,胞外壳寡糖的含量达20g/L以上。
虽然本发明已以较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,因此本发明的保护范围应该以权利要求书所界定的为准。