一种基因网络表达状态的二值化观测方法及系统
文献发布时间:2023-06-19 19:30:30
技术领域
本发明涉及生命科学领域的细胞基因表达量测定技术领域,特别是涉及一种基因网络表达状态的二值化观测方法及系统。
背景技术
随着生命科学技术的进步,基于基因表达量对细胞状态进行研究,能够得到更加精确的数据结果。目前存在的获取基因表达量的常见方法是通过基因测序匹配细胞中所有的RNA序列,进而统计出相同的基因片段,通过数据处理得到基因的表达量;此外,常用的还有PCR,qPCR,数字PCR的方法,通过对细胞内的全部RNA进行测量,最终获得相对定量或绝对定量的基因表达量。
在使用基因测序(NGS等)的方式获取基因表达量的过程中,需要使用特定的仪器或将样本邮寄到公司测量;并且,获得的测序数据需要通过算法转换为对应的基因表达量。而上述过程中,获取基因表达量将花费数周时间,具有较高的成本。在使用常用的PCR和qPCR仪器时,一般只能配备96孔板或384孔板,传统基因测序和PCR方法均无法在使用单个孔板进行实验,即无法在一台PCR仪器中同时获得基因表达谱所需基因维度的基因数据,大大提升了实验的时间和成本。
同时,目前存在一些对于基因进行降维观测的理论方式,可以快速获取细胞中部分基因的表达量,如神经网络降维方式,通过部分基因测量结合早期分析数据和生物学关系,推断剩余基因表达谱等。上述方式可以在理论层面上降低采样率,但是目前存在的降维方式,均存在数据可信度低、设计试验方案流程复杂、无法同时保留基因数据的线性信息与非线性信息、成本较高等问题,限制了上述方法在实际中的应用场景。
发明内容
本发明的目的是提供一种基因网络表达状态的二值化观测方法及系统,降低基因表达测量出错率,提高基因表达量的测量稳定性。
为实现上述目的,本发明提供了如下方案:
一种基因网络表达状态的二值化观测方法,包括:
根据基因字典和被测细胞的基因维度,确定随机测量矩阵;所述随机测量矩阵包括0值、正值和负值;所述基因字典是对样本集进行字典训练得到的;所述样本集包括多个样本;每个所述样本包括样本细胞的多维度基因测序数据;
根据所述随机测量矩阵和所述被测细胞的基因维度,确定所述随机测量矩阵对应的多组基因名称组;每组所述基因名称组包括正值基因名称小组和负值基因名称小组;所述基因名称组的组数量与所述随机测量矩阵的行数相同;
针对所述随机测量矩阵对应的每组基因名称组,采用T7分别定制正值引物和负值引物;所述正值引物对应所述正值基因名称小组,所述负值引物对应所述负值基因名称小组;
对所述正值引物和所述负值引物分别进行标记,以得到对应的正值标记引物和负值标记引物;
采用所述正值标记引物对所述正值基因名称小组对应的基因数据进行扩增,采用所述负值标记引物对所述负值基因名称小组对应的基因数据进行扩增,以得到综合基因扩增数据;
对所述综合基因扩增数据进行标记观测,以得到基因观测数据;所述基因观测数据包括正值观测数据和负值观测数据;
根据所述基因观测数据和所述基因字典,计算所述被测细胞的基因表达量。
可选地,所述根据基因字典和被测细胞的基因维度,确定随机测量矩阵,具体包括:
根据预设稀疏度和被测细胞的基因维度,构建初步随机矩阵;
根据被测细胞的基因维度确定被测细胞的多维度基因测序数据;
将所述被测细胞的多维度基因测序数据输入至基因字典,以得到稀疏参考测量矩阵;
将所述初步随机矩阵与所述稀疏参考测量矩阵进行偏差计算,得到偏差结果;
当所述偏差结果未处于预设偏差范围内时,返回根据预设稀疏度和被测细胞的基因维度,构建初步随机矩阵的步骤;
当所述偏差结果处于预设偏差范围内时,所述初步随机矩阵为随机测量矩阵。
可选地,所述根据预设稀疏度和被测细胞的基因维度,构建初步随机矩阵,具体包括:
基于有限等距条件,根据预设稀疏度和被测细胞的基因维度,计算基因整体采样率和基因单行采样率;
根据所述基因整体采样率和所述基因单行采样率,生成初步随机矩阵。
可选地,所述基因整体采样率的计算公式为:
,或,/>
所述基因单行采样率的计算公式为:
;
其中,
可选地,所述基因字典的训练过程,具体包括:
获取样本集和当前迭代次数;
根据所述样本集,随机生成初级基因字典矩阵和初级活跃度矩阵;
对所述初级基因字典矩阵依次进行迭代计算和标准化处理,以得到次级基因字典矩阵;
采用正交匹配跟踪算法,对所述初级活跃度矩阵进行迭代计算,以得到次级活跃度矩阵;
将所述当前迭代次数更新为当前迭代次数+1;
判断所述次级基因字典矩阵和所述次级活跃度矩阵是否满足预设迭代结束条件,以及判断所述当前迭代次数是否达到预设迭代次数;
当所述次级基因字典矩阵和所述次级活跃度矩阵满足预设迭代结束条件,或者,所述当前迭代次数达到预设迭代次数时,输出所述次级基因字典矩阵和所述次级活跃度矩阵;所述次级基因字典矩阵和所述次级活跃度矩阵构成基因字典;
当所述次级基因字典矩阵和所述次级活跃度矩阵不满足预设迭代结束条件,并且,所述当前迭代次数未达到预设迭代次数时,返回根据所述样本集,随机生成初级基因字典矩阵和初级活跃度矩阵的步骤。
可选地,对初级基因字典矩阵进行迭代计算的公式为:
;
其中,U1表示迭代后的初级基因字典矩阵,U表示初级基因字典矩阵,
可选地,所述采用正交匹配跟踪算法,对所述初级活跃度矩阵进行迭代计算,以得到次级活跃度矩阵,具体包括:
确定正交匹配跟踪算法的参数初始值;所述参数初始值包括初始残差、支撑索引集和迭代初始值;
根据所述初始残差和所述初级活跃度矩阵,确定最大相关的索引;
将所述最大相关的索引加入至所述支撑索引集,以得到更新后的支撑索引集;
根据所述更新后的支撑索引集和所述初级活跃度矩阵,对所述初始残差进行更新;
将所述迭代初始值更新为迭代初始值加一;
判断更新后的所述迭代初始值是否达到预设值;
当更新后的所述迭代初始值未达到预设值时,返回根据所述初始残差和所述初级活跃度矩阵,确定最大相关的索引的步骤;
当更新后的所述迭代初始值达到预设值时,根据更新后的所述初始残差计算稀疏系数,并输出所述稀疏系数和所述更新后的支撑索引集;所述稀疏系数和所述更新后的支撑索引集构成次级活跃度矩阵。
可选地,对所述正值引物和所述负值引物分别进行标记,具体包括:
对所述正值引物和所述负值引物,采用不同的荧光染料进行染色;
对所述综合基因扩增数据进行标记观测,以得到基因观测数据,具体包括:
采用荧光检测装置,检测所述综合基因扩增数据中荧光染料的发光强度;
根据所述荧光染料的发光强度将观测到的基因数据划分为正值观测数据和负值观测数据。
可选地,根据所述基因观测数据和所述基因字典,计算所述被测细胞的基因表达量,具体包括:
根据所述基因观测数据和所述基因字典,计算基因活跃度矩阵;
根据所述基因活跃度矩阵和所述基因字典,计算初步基因表达量;所述初步基因表达量包括正基因表达量和负基因表达量;
将所述初步基因表达量中的负基因表达量作为缺失值处理,以得到所述被测细胞的基因表达量。
为达上述目的,本发明还提供了如下技术方案:
一种基因网络表达状态的二值化观测系统,包括:
随机矩阵生成模块,用于根据基因字典和被测细胞的基因维度,确定随机测量矩阵;所述随机测量矩阵包括0值、正值和负值;所述基因字典是采用对样本集进行字典训练得到的;所述样本集包括多个样本;每个所述样本包括样本细胞的多维度基因测序数据;
基因名称二值化区分模块,用于根据所述随机测量矩阵和所述被测细胞的基因维度,确定所述随机测量矩阵对应的多组基因名称组;每组所述基因名称组包括正值基因名称小组和负值基因名称小组;所述基因名称组的组数量与所述随机测量矩阵的行数相同;
引物生成模块,用于针对所述随机测量矩阵对应的每组基因名称组,采用T7分别定制正值引物和负值引物;所述正值引物对应所述正值基因名称小组,所述负值引物对应所述负值基因名称小组;
引物标记模块,用于对所述正值引物和所述负值引物分别进行标记,以得到对应的正值标记引物和负值标记引物;
基因扩增模块,用于采用所述正值标记引物对所述正值基因名称小组对应的基因数据进行扩增,采用所述负值标记引物对所述负值基因名称小组对应的基因数据进行扩增,以得到综合基因扩增数据;
基因观测模块,用于对所述综合基因扩增数据进行标记观测,以得到基因观测数据;所述基因观测数据包括正值观测数据和负值观测数据;
基因表达量计算模块,用于根据所述基因观测数据和所述基因字典,计算所述被测细胞的基因表达量。
根据本发明提供的具体实施例,本发明公开了以下技术效果:
本发明提供了一种基因网络表达状态的二值化观测方法及系统,构建基因字典,根据基因字典和被测细胞的基因维度,确定随机测量矩阵,并且随机测量矩阵中仅包括0值、正值和负值。根据随机测量矩阵和被测细胞的基因维度,确定随机测量矩阵对应的多组基因名称组,以实现对于基因名称的二值化格式的设计。根据随机测量矩阵对应的基因名称组,采用T7分别定制正值引物和负值引物;对正值引物和负值引物分别进行标记,以得到对应的正值标记引物和负值标记引物;采用正值标记引物对正值基因名称小组对应的基因数据进行扩增,采用负值标记引物对负值基因名称小组对应的基因数据进行扩增,以得到综合基因扩增数据。由于本发明采用常见的T7启动子方式进行基因扩增,能够使得该方法更具有普适性,降低了成本。进而进行标记观测,以得到基因观测数据。其中,基因观测数据中保留了对应观测数据的符号信息,比如正负,从而实现了对观测数据的二值化观测。最后根据二值化的基因观测数据和基因字典,计算被测细胞的基因表达量,通过正负的二值化观测数据能够减少测量过程中某一错误数据对最终结果的影响,提高测量的稳定性。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其他的附图。
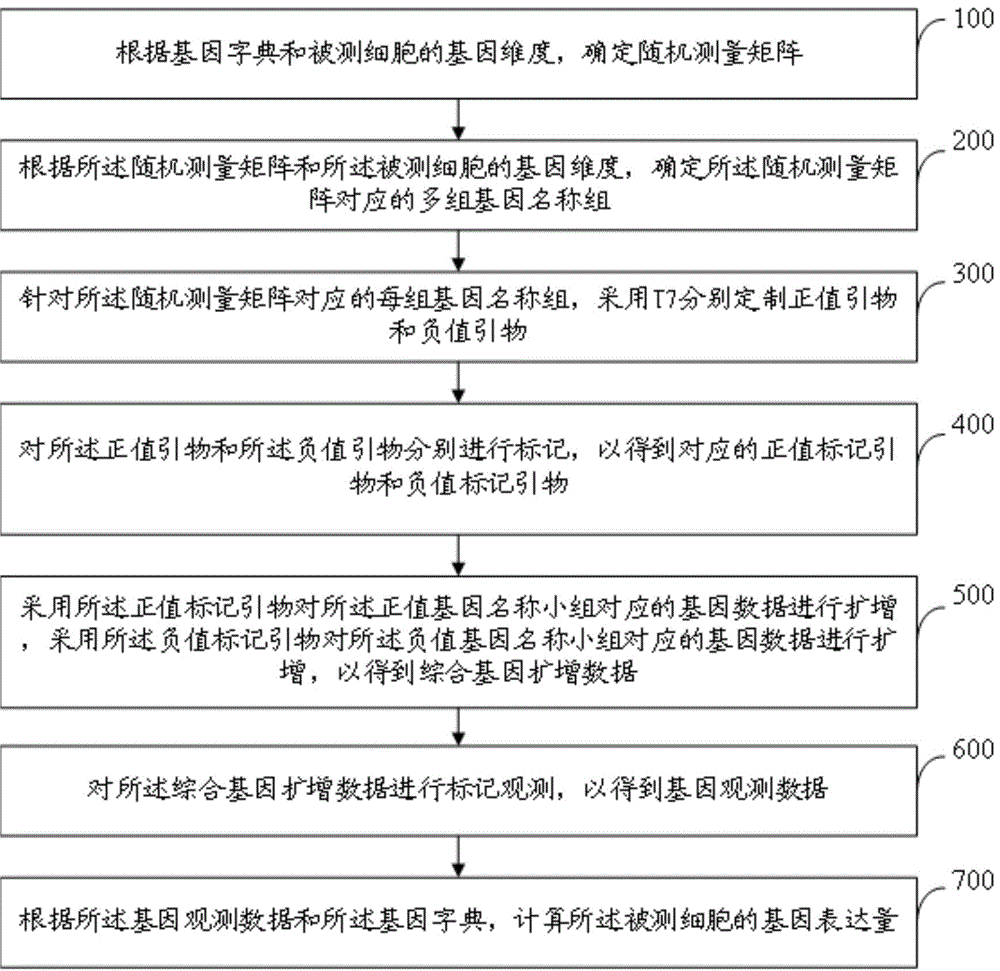
图1为本发明基因网络表达状态的二值化观测方法的流程示意图;
图2为本发明基因网络表达状态的二值化观测系统的结构示意图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明提供一种基因网络表达状态的二值化观测方法及系统,基于RIP条件(压缩感知中观测矩阵的有限等距性质,Restricted Isometry Property)对基因进行稀疏编码,使用线性扩增方式实现对降维数据的观测过程,通过公开基因测序数据集训练基因字典,通过实验方法获取观测数值,使用压缩感知算法重建细胞的基因表达量,最终达到降低实验成本,缩短测量时间的要求。
为使本发明的目的、特征和优点能够更加明显易懂,下面结合附图和具体实施方式对本发明作进一步详细的说明。
实施例一
如图1所示,本实施例提供一种基因网络表达状态的二值化观测方法,包括:
步骤100,根据基因字典和被测细胞的基因维度,确定随机测量矩阵;所述随机测量矩阵包括0值、正值和负值;所述基因字典是对样本集进行字典训练得到的;所述样本集包括多个样本;每个所述样本包括样本细胞的多维度基因测序数据。在实际应用中,为了便于计算和减少计算难度,正值和负值一般通过+1与-1进行设置和确定。
步骤100具体包括:
1)根据预设稀疏度和被测细胞的基因维度,构建初步随机矩阵。
具体地,基于有限等距条件(RIP条件),根据预设稀疏度K和被测细胞的基因维度n,计算基因整体采样率和基因单行采样率;然后,根据所述基因整体采样率和所述基因单行采样率,通过预设的随机算法,生成初步随机矩阵。
基因的稀疏度与基因维度有关,对于人总体的20000基因来说,一般将预设稀疏度设置为K=10-15,所述基因整体采样率的计算公式为:
其中,
2)根据被测细胞的基因维度确定被测细胞的多维度基因测序数据。
3)将所述被测细胞的多维度基因测序数据输入至基因字典,以得到稀疏参考测量矩阵。
4)将所述初步随机矩阵与所述稀疏参考测量矩阵进行偏差计算,得到偏差结果。当所述偏差结果未处于预设偏差范围内时,返回步骤1),并调节s的值,以重新构建初步随机矩阵;当所述偏差结果处于预设偏差范围内时,所述初步随机矩阵为随机测量矩阵。
在实际应用中,将被测细胞的多维度基因测序数据输入至训练好的基因字典之后,可直接通过人为观察基因输出的稀疏参考测量矩阵与初步随机矩阵,得到稀疏参考测量矩阵相对于初步随机矩阵的复现效果,进而人为得出是否需要重新构建初步随机矩阵的结论。
在基因字典的构建方面,通过SMAF算法进行字典训练,SMAF算法获取的字典更加稀疏,可以被提高单比特压缩感知的数据记录方式复现出的数据精确性。所述基因字典的训练过程,具体包括:
1)获取样本集和当前迭代次数。样本集中多个样本的基因维度相同,且样本集中各个样本的基因测序数据是已知公开的。
2)根据所述样本集,随机生成初级基因字典矩阵U(m
3)对所述初级基因字典矩阵依次进行迭代计算和标准化处理,以得到次级基因字典矩阵。
具体地,使用非负参数推断(LassoNonnegative)进行字典迭代过程,其公式如下:
;
可转换为如下公式:
;
其中,U1表示迭代后的初级基因字典矩阵,U表示初级基因字典矩阵,
对迭代后的初级基因字典矩阵进行标准化处理,使得
4)采用正交匹配跟踪算法((Orthogonal Matching Pursuit,OMP算法),对所述初级活跃度矩阵进行迭代计算,以得到次级活跃度矩阵。
5)将所述当前迭代次数更新为当前迭代次数+1。
6)判断所述次级基因字典矩阵和所述次级活跃度矩阵是否满足预设迭代结束条件,以及判断所述当前迭代次数是否达到预设迭代次数。当所述次级基因字典矩阵和所述次级活跃度矩阵满足预设迭代结束条件,或者,所述当前迭代次数达到预设迭代次数时,输出所述次级基因字典矩阵和所述次级活跃度矩阵;所述次级基因字典矩阵和所述次级活跃度矩阵构成基因字典。当所述次级基因字典矩阵和所述次级活跃度矩阵不满足预设迭代结束条件,并且,所述当前迭代次数未达到预设迭代次数时,返回上文的步骤2),再次生成随机的初级基因字典矩阵U(m
在一个具体实际应用中,预设迭代次数为10次,预设迭代结束条件为x=U
其中,上文中步骤4)所述采用正交匹配跟踪算法,对所述初级活跃度矩阵进行迭代计算,以得到次级活跃度矩阵,具体包括:
A)确定正交匹配跟踪算法的参数初始值;所述参数初始值包括初始残差、支撑索引集和迭代初始值d。
B)根据所述初始残差和所述初级活跃度矩阵,确定最大相关的索引;具体地,最大相关的索引
;
其中,初级活跃度矩阵C=[c
C)将所述最大相关的索引加入至所述支撑索引集
;
D)根据所述更新后的支撑索引集和所述初级活跃度矩阵,对所述初始残差进行更新;具体地,初始残差的更新公式为:
;
其中,x是样本集X中的样本的基因测序数据,
E)将所述迭代初始值更新为迭代初始值加一;d=d+1。
F)判断更新后的所述迭代初始值是否达到预设值D;当更新后的所述迭代初始值未达到预设值时,返回步骤B);当更新后的所述迭代初始值达到预设值时,根据更新后的所述初始残差计算稀疏系数,并输出所述稀疏系数和所述更新后的支撑索引集;所述稀疏系数和所述更新后的支撑索引集构成次级活跃度矩阵。其中,更新后的支撑索引集
;
步骤200,根据所述随机测量矩阵和所述被测细胞的基因维度,确定所述随机测量矩阵对应的多组基因名称组;每组所述基因名称组包括正值基因名称小组和负值基因名称小组;所述基因名称组的组数量与所述随机测量矩阵的行数相同。
步骤300,针对所述随机测量矩阵对应的每组基因名称组,采用T7分别定制正值引物和负值引物;所述正值引物对应所述正值基因名称小组,所述负值引物对应所述负值基因名称小组。具体地,根据随机测量矩阵中每行基因数据对应的两组基因组合,分别制备被T7启动子包被的基因引物,即T7引物,得到正值T7引物小组(正值引物)和负值T7引物小组(负值引物)。
步骤400,对所述正值引物和所述负值引物分别进行标记,以得到对应的正值标记引物和负值标记引物。具体地,将所述正值基因名称小组对应的T7引物、所述负值基因名称小组对应的T7引物分别进行标记,以得到对应的正值标记引物和负值标记引物。优选地,将正值T7引物小组,采用第一荧光染料进行染色,得到正值标记引物;将负值T7引物小组,采用第二荧光染料进行染色,得到负值标记引物。其中,第一荧光染料与第二荧光染料的颜色不同。
步骤500,提取被测细胞中的RNA,采用所述正值标记引物对所述正值基因名称小组对应的基因数据进行扩增,采用所述负值标记引物对所述负值基因名称小组对应的基因数据进行扩增,以得到综合基因扩增数据。
具体地,无论是对正值基因名称小组进行的扩增,还是对负值基因名称小组进行的扩增,均为线性扩增,此过程中会将需要被设计的基因引物维度降低一半,单比特的方式需要提升被测基因的个数,线性扩增的方式可以有效的提升设计引物的特异性,更适用于单比特压缩感知。
步骤600,对所述综合基因扩增数据进行标记观测,以得到基因观测数据;所述基因观测数据包括正值观测数据和负值观测数据。
具体地,在线性扩增结束后,采用荧光检测装置,检测所述综合基因扩增数据中荧光染料的发光强度;根据所述荧光染料的发光强度将观测到的基因数据划分为正值观测数据和负值观测数据。即检测第一荧光染料和第二荧光染料的发光强度,可对应的记录为不同符号的数据,具体的正负的符号与亮度较大的颜色所对应的符号相同。
步骤700,根据所述基因观测数据和所述基因字典,计算所述被测细胞的基因表达量。优选地,步骤700包括:
1)根据所述基因观测数据和所述基因字典,计算基因活跃度矩阵;具体地,根据公式
2)根据所述基因活跃度矩阵和所述基因字典,计算初步基因表达量;所述初步基因表达量包括正基因表达量和负基因表达量;具体地,根据公式
3)将所述初步基因表达量中的负基因表达量作为缺失值处理,以得到所述被测细胞的基因表达量。
综上可知,本发明基因网络表达状态的二值化观测方法,为一种细胞基因表达量的快速获取方式,基于基因的模块化表达和具有可被压缩的性质,使用复合测量的方式,对基因进行稀疏编码,通过PCR反应实现对基因数据的降维观测,之后结合基因字典得出基因表达量。这种方法可以使用常见的PCR仪器或qPCR仪器进行测量,后续仍然提供了磁珠、微阵列、T7启动子等常见的基因扩增方式搭配压缩感知等算法用以获取基因表达谱,最终实现低成本(约为NGS的10%),高效率(单次测量时间约为2h),较高精确度(皮尔逊相关系数70%以上,皮尔逊相关系数65%以上),对实验精度要求较低,且可以去除批次效应的方式测量生物体的基因表达量。
另外,在实际应用方面,通过研究发现,对于同一种类型的疾病,其基因间的相互作用均与其表达量具有很强的相关性,而相同的基因编码方式(即同一个测量矩阵)固定的情况下,对基因的相互作用关系进行压缩获取,进而可以复现出基因表达谱。基于此,本发明使用开源数据对基因建立字典,之后通过稀疏编码的测量矩阵确定被测量基因,使用多重PCR反应(real-time PCR,qPCR等)获取基因的符号观测值,并仅通过符号观测值,基于压缩感知算法解码基因实现对细胞基因表达量的重建,获取当前细胞状态,整体操作可在2h内获取当前细胞的基因表达量,同时这一成本约为现有第二代基因测序(NGS)的10%。并且,其为重大疾病如:癌症,慢性病,重大传染病的治疗、预后以及病理分析提供基因层级上的数据指导。要知道,对于疾病治疗的最优方式是通过药物调节细胞中基因表达量,进行基因层面上的治疗,从而实现疾病治疗,所以快速低成本的获取细胞的基因表达量是实现上述方法的关键技术。
实施例二
如图2所示,为了执行上述实施例一对应的方法,以实现相应的功能和技术效果,本实施例提供了一种基因网络表达状态的二值化观测系统,包括:
随机矩阵生成模块101,用于根据基因字典和被测细胞的基因维度,确定随机测量矩阵;所述随机测量矩阵包括0值、正值和负值;所述基因字典是采用对样本集进行字典训练得到的;所述样本集包括多个样本;每个所述样本包括样本细胞的多维度基因测序数据。
基因名称二值化区分模块201,用于根据所述随机测量矩阵和所述被测细胞的基因维度,确定所述随机测量矩阵对应的多组基因名称组;每组所述基因名称组包括正值基因名称小组和负值基因名称小组;所述基因名称组的组数量与所述随机测量矩阵的行数相同。
引物生成模块301,用于针对所述随机测量矩阵对应的每组基因名称组,采用T7分别定制正值引物和负值引物;所述正值引物对应所述正值基因名称小组,所述负值引物对应所述负值基因名称小组。
引物标记模块401,用于对所述正值引物和所述负值引物分别进行标记,以得到对应的正值标记引物和负值标记引物。
基因扩增模块501,用于采用所述正值标记引物对所述正值基因名称小组对应的基因数据进行扩增,采用所述负值标记引物对所述负值基因名称小组对应的基因数据进行扩增,以得到综合基因扩增数据。
基因观测模块601,用于对所述综合基因扩增数据进行标记观测,以得到基因观测数据;所述基因观测数据包括正值观测数据和负值观测数据。
基因表达量计算模块701,用于根据所述基因观测数据和所述基因字典,计算所述被测细胞的基因表达量。
现对于现有技术,本发明还具有如下优点:
(1)本发明将对基因数据降维的理论方式与实际生化反应相结合,具体地,将RIP条件与基因扩增的实际方法(T7启动子)进行反应结合,将理论降维方式通过生化反应进行实现,把基因高维信息通过多重PCR的方式进行储存;并且此种方式无需特殊仪器进行操作,实验过程中仅需要使用T7启动子线性扩增反应即可达到目的,提供了一种稳定且普适性强的获取基因表达量的方式。
(2)本发明提供了短时、精确且成本低的细胞基因表达量的获取方法,将基因进行稀疏编码并结合压缩感知的字典训练方法,获取细胞基因表达量,单次实验成本约为第二代基因测序(NGS)的10%,实验时间为2h,约为NGS的1%,测量精度较高(皮尔逊相关系数70%以上,斯皮尔曼相关系数65%以上)。
(3)本发明提供的基因表达量测量方式,可以避免测序过程中的批次效应与测序深度的影响,做到将每一次的测量获得的基因表达量都拥有去批次效果,大幅减少后续处理数据工作量。
本说明书中各个实施例采用递进的方式描述,每个实施例重点说明的都是与其他实施例的不同之处,各个实施例之间相同相似部分互相参见即可。
本文中应用了具体个例对本发明的原理及实施方式进行了阐述,以上实施例的说明只是用于帮助理解本发明的方法及其核心思想;同时,对于本领域的一般技术人员,依据本发明的思想,在具体实施方式及应用范围上均会有改变之处。综上所述,本说明书内容不应理解为对本发明的限制。
- 一种基于异步状态观测器的马尔科夫跳变系统控制方法
- 一种基于自适应状态观测器的网络化运动控制系统状态估计方法
- 热休克诱导外源基因高效表达的蓝藻转基因表达系统及其用于表达胸腺素α1的方法