一种TMT项目图谱解析和数据分析的方法及系统
文献发布时间:2023-06-19 19:30:30
技术领域
本发明属于生物医药技术领域,涉及一种串联质谱标签TMT项目图谱解析和数据分析方法及系统。
背景技术
目前,在企业生产中,存在大量TMT项目谱图解析和数据分析需求,以往这些工作都是通过人工手动的方式进行处理的,步骤很多,操作繁琐。首先,分析人员要筛选出需要进行谱图解析的项目,找到该项目的原始数据路径和蛋白数据库路径,需要和实验人员确定项目的标记定量方法,然后手动将这些谱图解析所需的文件添加到谱图解析软件中,接着手动设置软件的相关参数并开始谱图解析,由于谱图解析过程耗时较久,且不同项目耗时不同,需要分析人员人工查看解析过程是否完成,以便开启下一步数据分析工作。在数据分析时,需要分析人员手动输入相关信息到数据分析软件中。最后,在项目完成数据分析后,需要手动整理分析结果。整个过程,极其耗费时间和精力,需要较多的分析人员协同合作来完成该项工作,人力成本很高。
发明内容
本发明的目的是提供一种TMT项目谱图解析和数据分析方法,可以实现从谱图解析所需文件的定位,文件导入谱图分析软件、谱图解析过程的监督,谱图解析结果数据分析的全自动化操作,极大的解决了目前TMT项目谱图解析和数据分析中操作繁琐、周期长、人力成本高等问题。
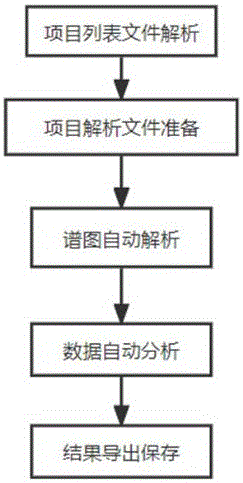
本发明提出了一种TMT项目谱图解析和数据分析方法,所述方法包括如下具体步骤:
步骤一、项目列表文件解析
步骤1.1、利用python读取TMT项目的项目列表文件,遍历项目列表文件中的每一行文字;
步骤1.2、将每一行包含“上机”或“搜库结题”字段,且同时包含“TMT”字段的记录放入python列表中。
步骤1.1中,所述项目列表文件是一个文本文件或excel表格文件,其中每一行文字记录了一个项目的一种谱图解析和数据分析任务,且每一行包含项目合同号、项目类型和样本类型信息;
步骤1.2中,所述“TMT”是指项目的类型,所述“上机”和“搜库结题”可以用来区分样本类型,“上机”说明项目需要对质控样本进行谱图解析和数据分析,“搜库结题”说明需要对正式样本进行谱图解析和数据分析。
步骤二、项目解析文件准备
步骤2.1中,根据合同号,使用get_pro_db_bjsx方法获取项目的标记信息文件和蛋白数据库文件的绝对路径。/>
所述绝对路径是指以根目录为起点能够直接到达某一个目录的路径,准确地说是某些文件或者目录。
步骤2.2中,根据合同号,使用获取原始文件路径get_rawdata_path方法获取预设的项目原始数据文件的绝对路径和谱图解析结果输出目录的绝对路径,即原始数据文件的具体存储位置和结果文件存放的具体位置。
步骤2.3中,根据标记信息文件和定量模板文件,使用mk_qm_method方法生成项目的定量方法文件。
步骤2.1中所述get_pro_db_bjsx方法,为本发明中利用python语言自行研发的方法,可以根据项目的合同号判断项目的标记信息文件、蛋白数据库文件是否存在,准确提供项目标记信息文件和蛋白数据库文件的绝对路径。通过合同号的字符串中第3-6位的数字确定合同年份、通过合同号的字符串中第3-8位数字确定合同的月份信息,方法会在合同号中年份和月份对应的文件夹中进行文件检索,而忽略其他年份和该年份其他月份对应的文件夹,当找到对应年份的对应月份文件夹后,使用os.listdir函数遍历合同对应年份和月份所在文件夹下所有文件夹,并确定名称中包含该合同号的文件夹作为项目的文件夹(例如合同号MJ20220802134中,年份位2022,月份位202208,项目所在路径为J:实验生产部蛋白实验数据2022202208MJ20220802134-冯人伟-刘梓清-TMT-6例组织样品),该方法会在项目所在文件夹的“生产附件”子文件夹中通过os.walk遍历所有文件,根据“标记.xls”锁定该项目的标记信息文件,该方法会在“数据库”子文件夹中遍历所有文件,并以“fasta”为后缀为标准定位蛋白数据库。
步骤2.1中所述的标记信息文件是一个excel表格,第一行为表头信息,第二行的第一列为项目使用的标记试剂批号,其余列为项目使用的标记信息,如图3所示。
步骤2.1中所述的蛋白数据库文件是一个以“fasta”为后缀的文件,文件中包含了蛋白序列号、注释信息和蛋白序列,如图4所示。
步骤2.2中所述get_rawdata_path方法,为本发明中利用python语言自行研发的方法,可以根据项目合同号判断项目原始数据文件是否存在,并过滤干扰信息,严格区分质控样本的原始数据文件和正式样本的原始数据文件,准确提供项目原始数据文件的绝对路径;提供质控样本谱图解析结果和正式样本谱图解析结果存放的绝对路径。
步骤2.2中所述的原始数据文件是以“raw”为后缀的文件,是质谱下机数据。所述输出目录是指谱图解析产生结果文件存放的位置,不同项目的谱图解析结果需要存放在不同的位置。
步骤2.3中所述mk_qm_method方法,为本发明中利用python语言自行研发的方法,可以根据标记信息文件生成谱图解析所需的标记定量文件,并将其移动到正确的位置。所述mk_qm_method方法利用python语言,根据以下内容实现:
步骤2.3中所述的定量模板文件是一个后缀为“method”的文件,文件中内容为XML格式,为本发明特制文件。在本发明中不同批号的标记试剂对应不同的定量模板文件,例如批号为“WH324722”的标记试剂对应“TMTpro 16PlexLOT_WH324722.method”定量模板文件,批号为“WJ325156”的试剂对应“TMTPro 16plexLOT_WJ325156.method”定量模板文件,批号为“XE354084”的标记试剂对应“TMTPro 16PlexLOT_XE354084.method”定量模板文件。不同批号的定量模板文件内容格式相同,模板中的校正因子不同,不同定量模板的校正因子在制作模板时已正确设定;模板中的校正因子用于校正不同标记试剂定量系数。定量系数是从标记试剂的说明书中获取的,在定量的过程中需要使用到,本发明对定量系数本身并未作改变,完全按照说明书的数值来的。只是在定量过程中需要把定量系数添加到软件中,这个添加过程,本发明进行了一些优化。常规的方法需要对照说明书,将不同标记的定量系数一个接着一个的添加到软件中,当使用的标记数目不同时,还需要对标记进行增删,几乎一个项目需要手动操作一次,异常繁琐,费时费力,且容易出错。本发明制作了定量模板,把定量系数添加到模板中,只进行了一次手动操作。后续只需使用程序调用模板即可,在面对不同项目使用不同标记的情况时候,程序会自动从定量模板中调取定量系数,并对标记进行增删,完全不需要人手动操作,省时省力,几乎不出错。
步骤2.3中,本发明特制的不同定量模板文件中均包含了16个标记(126、127N、127C、128N、128C、129N、129C、130N、130C、131N、131C、132N、132C、133N、133C、134N)的相关信息,每一个标记的众多属性中均包含“name="IsActive"”属性,当某一个标记该属性的值为“True”时,说明该标记在项目中有使用,当某一个标记该属性的值为“False”时,说明项目没有使用该标记,通过修改不同标记该属性的值来表示本项目最终使用的标记。本发明不同定量模板文件中不同标记的“name="IsActive"”属性的值均设置为“%s”。
步骤2.3中所述mk_qm_method方法基本实现过程为,首先利用pandas.read_csv或者pandas.read_excel函数,从标记信息文件中解析出项目使用的标记试剂批号和项目使用的标记,根据标记试剂批号和定量模板文件的对应关系确定项目的定量模板文件,并利用python中with open方法读取该定量模板文件,并作为字符串存储在内存空间中。根据从标记信息文件中解析得到的项目使用标记,生成一个包含16个元素的python列表,列表中的元素依次对应16个标记,代表标记是否在项目中使用与否,如果标记在项目中使用,则在python列表中对应位置的元素值为“True”,否则,对应位置的元素值为“False”。使用python中格式化的方法将定量模板文件中不同标记的“name="IsActive"”属性值直接替换成python列表中的元素值,即可得到项目的定量方法文件。
步骤2.3中所述定量方法文件,记录了项目使用的标记信息和校正因子信息,是谱图解析过程中需要用到的文件。
步骤三、谱图自动解析
步骤3.1、使用pd_start方法,启动谱图解析软件,并进入管理界面。
步骤3.2、使用add_db方法,删除谱图解析软件中已有的蛋白数据库,导入项目对应蛋白数据库文件。
步骤3.3、使用add_method方法,删除谱图解析软件中已有的定量方法,导入项目对应的定量方法文件。
步骤3.4、使用outdir_select方法,选择谱图解析结果保存的位置。
步骤3.5、使用workflow_select方法,选择项目的工作流程(包括processingworkflow和consensus workflow)。
步骤3.6、使用qm_select方法,选择项目的定量方法。
步骤3.7、使用rawdata_import方法将项目原始数据据文件导入谱图解析软件。
步骤3.8、使用parameter_set方法,对关键参数进行设定,并开启谱图解析过程。/>
步骤3.1-3.8中,所述pd_start方法、add_db方法、add_method方法、outdir_select方法、workflow_select方法、rawdata_import、parameter_set方法均为利用python语言为谱图解析特定研发,具有较强的特异性和稳定性,可以提高谱图解析过程中操作的准确性,有效的保障谱图解析过程的稳定进行。
上述步骤3.1-3.8中应用的函数方法利用python进行开发,在通过python进行开发的过程中,上述函数方法的逻辑结构由本发明通过实际情况进行设定。
pd_start方法:通过pywinauto中application.Application函数来链接pd软件,并通过start函数启动pd软件;由于软件的启动受到多种因素的影响,而且只有软件完全正常的启动之后,才能够采取下一步操作,因此需要对软件启动状态进行判断,通过pyautogui.locateOnScreen函数每隔1秒在整个屏幕上捕获pd软件的设置按钮(见图8中的a处),当捕获到该图像时说明软件已经正常启动,否则,软件还没有正常启动,使用time.sleep函数进行等待。如果pd软件超过2分钟仍未成功启动,则通过os.system函数和taskkill命令将软件进程终止,然后再次通过pywinauto中application.Application函数来链接pd软件,并通过start函数重新启动pd软件。如果重新启动pd软件3次均未能成功,则通过DingtalkChatbot通知分析人员该信息,以便及时排除相关问题。该方法有效的提高了流程运行的稳定性,有效的降低了各种偶然因素(比如电脑状况)对系统的影响。
add_db方法、add_method方法、outdir_select方法、workflow_select方法、qm_select方法、rawdata_import方法、parameter_set方法具有一定的相似性,即都是使用pyautogui进行图像识别和模拟鼠标键盘操作,只是应用场景不同。
步骤3.1中所述的谱图解析软件是指'Thermo Proteome Discoverer 2.4',是一种功能强大的蛋白质组学数据谱图解析软件。
步骤3.1中所述pd_start方法首先通过python启动谱图解析软件以代替传统手动点击方式启动软件,软件启动时间受制于计算机性能和状态等诸多因素,存在较大的不稳定性,是自动进行谱图解析过程的重大障碍,本方法通过python对软件界面中“设置”按钮进行图像识别来判断软件是否启动,每1秒进行一次图像捕获,如果该按钮出现在电脑屏幕中,则说明软件已经启动,可以开始下一步操作,否则说明软件仍在启动之中,仍需继续等待。
步骤3.1中所述进入管理界面,是通过get_position方法获取谱图解析软件中“设置”按钮在屏幕中的坐标,通过pyautogui.leftClick点击该坐标,进入管理界面。
所述get_position方法是对pyautogui.locateOnScreen的优化,可以按照图像查找精确度confidence由高到低的次序依次进行图像捕获直到找到图像在屏幕中的位置为止,增加了图像识别的灵活性,提高了图像识别的稳定性。
步骤3.2中所述add_db方法首先通过get_position方法获取谱图解析软件中蛋白数据库管理按钮在屏幕中的坐标,通过pyautogui.leftClick点击该坐标,进入蛋白数据库管理界面,通过get_position方法获取谱图解析软件中数据库删除按钮在屏幕中的坐标,通过pyautogui.leftClick点击该坐标,删除软件中已有的蛋白数据库,并通过get_position方法获取谱图解析软件中数据库导入按钮在屏幕中的坐标,通过pyautogui.leftClick点击该坐标,导入项目的蛋白数据库文件。
步骤3.3中所述add_method方法,通过get_position方法获取谱图解析软件中定量方法删除按钮在屏幕中的位置,通过pyautogui.leftClick点击该坐标,删除软件中已有的标记定量方法,通过get_position方法获取谱图解析软件中定量方法导入按钮在屏幕中的位置,通过pyautogui.leftClick点击该坐标,导入项目定量方法文件。
步骤3.4中所述outdir_select方法,通过get_position方法获取谱图解析软件中新建项目按钮在屏幕中的坐标,通过pyautogui.leftClick点击该坐标,打开新建项目窗口,通过get_position方法获取输出文件夹选择按钮,通过pywinauto.keyboard.send_key将谱图解析结果输出目录的绝对路径输入软件的文本框内。
步骤3.5中所述workflow_select方法,通过get_position方法获取处理工作流(processing workflow)选择按钮在屏幕中的坐标,通过pyautogui.leftClick点击该坐标,打开处理工作流选择窗口,通过pywinauto.keyboard.send_key将处理工作流的绝对路径输入文本框,完成处理工作流的选择。通过get_position方法获取共识工作流(consensus workflow)选择按钮在屏幕中的坐标,通过pyautogui.leftClick点击该坐标,打开共识工作流选择窗口,通过pywinauto.keyboard.send_key将共识工作流的绝对路径输入文本框,完成共识工作流的选择。
步骤3.6所述qm_select方法,通过get_position方法获取软件中定量方法选择按钮在屏幕中的坐标,并通过pyautogui.leftClick点击该坐标,完成定量方法的设置。
步骤3.7中所述rawdata_import方法,通过get_position方法获取软件中原始数据导入按钮在屏幕中的坐标,并通过pyautogui.leftClick点击该坐标,打开原始数据选择窗口,pywinauto.keyboard.send_key将项目原始数据的绝对路径输入窗口的文本框中,完成原始数据的导入。
步骤3.8中所述parameter_set方法,通过get_position方法获取软件中参数修改按钮(Edit)在屏幕中的坐标,并通过pyautogui.leftClick点击该坐标,进入工作流(workflow)设置界面,通过get_position方法获取谱图文件重校正按钮(Spectrum FilesRC)在屏幕中的坐标,并通过pyautogui.leftClick点击该坐标,打开谱图文件重校正参数设置界面,通过get_position方法获取谱图解析软件中蛋白数据库选择按钮在屏幕中的位置,通过pyautogui.leftClick点击该坐标,完成蛋白数据库的设置。通过get_position方法获取谱图解析引擎(Sequest HT)在屏幕中的坐标,通过pyautogui.leftClick点击该坐标,打开谱图解析引擎参数设置界面,通过get_position方法获取蛋白数据库选择按钮在屏幕中的位置,通过pyautogui.leftClick点击该坐标,完成蛋白数据库的设置。通过get_position方法获取执行分析按钮(Run)在屏幕中的坐标,通过pyautogui.leftClick点击该坐标。通过get_position方法获取分析警告窗口中忽略按钮(Ignore)在屏幕中的位置,通过pyautogui.leftClick点击该坐标,开启谱图解析过程。
通过对质谱原始数据的解析,在谱图解析完成后能够获得不同样本中蛋白质的相对表达量,后续的差异分析或者成图是基于这些表达量信息进行。
步骤四、数据自动分析
步骤4.1、使用stat_check方法,每隔2-10分钟,对谱图解析结果输出目录中所有文件进行一次扫描,当结果输出目录中出现名称为“Protein_information.xls”的文件时,说明谱图解析过程已经完成且已经通过TMT_export方法生成谱图解析结果文件,即可开始数据分析步骤。/>
步骤4.2、在确认谱图解析过程完成后,利用python读取项目分组文件及对比文件、谱图解析产生的“protein.xls”文件。
步骤4.3、利用numpy和scipy对每一个蛋白按组别进行费歇尔精确检验,求出不同组别之间每个蛋白的差异倍数FC和Pvalue;
步骤4.4、利用sklearn和protein.xls文件中蛋白表达丰度的信息,按组别对样本进行主成分分析(PCA)。
步骤4.1中扫描间隔时间优选2分钟,可以在谱图解析完成后尽可能快的开始数据分析,减少时间消耗。
步骤4.1中所述的stat_check方法,是利用python语言自行研发,可以按照一定的时间间隔,对谱图解析结果输出目录进行扫描,获取谱图解析过程已产生的文件信息,包括文件名称、产生时间、文件大小,并可以判断谱图解析过程是否完成。
步骤4.1中所述的谱图解析结果文件,是使用TMT_export方法产生的4个表格文件,包括蛋白表格(protein.xls)、肽段表格(peptide.xls)、肽段匹配表格(psm.xls)、蛋白信息统计表格(Protein_information.xls)。
步骤4.1所述TMT_export方法,是利用python语言自行研发,可以镶嵌在谱图解析软件中,在谱图解析过程中,对相关解析信息进行整理,并输出为文件。
TMT_export方法:pd软件在进行谱图解析过程中生成了“proteins”、“ProteinGroups”、“Peptide Groups”、“PSMs”和“MS/MS Spectrum Info”等信息,但是这些信息无法自动导出。由于pd软件中内置了“Scripting Node”节点,通过该节点可以调取pd软件谱图解析过程中产生的相关信息,并利用python对信息进行处理。本发明设计了TMT_export方法并将其镶嵌在“Scripting Node”节点中,在谱图解析过程完成后,调取了“proteins”、“Protein Groups”、“Peptide Groups”、“PSMs”和“MS/MS Spectrum Info”等信息,并对其进行处理,产生了蛋白表格(protein.xls)、肽段表格(peptide.xls)、肽段匹配表格(psm.xls)、蛋白信息统计表格(Protein_information.xls)。
步骤4.1中所述的Protein_information.xls文件,记录了谱图解析结果中的相关信息,包括总谱图数目(Total Spectrum)、鉴定谱图数目(Identified Spectrum)、肽段数目(Peptide number)、蛋白数目(Protein number)和蛋白质组数目(Protein groupnumber)。
步骤4.2中所述分组文件和对比文件记录了谱图解析的每个样本所属的组别信息和不同组别之间的对比信息。
步骤4.3中所述费歇尔精确检验,是一种用来检验一次随机实验的结果是否支持对于某个随机实验的假设,当测试结果出现小概率事件则认定原有假设不被支持,理论源于超几何分布,以Pvalue作为检测值,计算的Pvalue越小,表示越远离零假设。
步骤4.3中所述FC和Pvalue,FC是蛋白表达丰度在组别间的差异倍数,Pvalue是差异的显著性标准,FC的阈值越大,Pvalue的阈值越小,筛选得到的差异表达蛋白数目越少;FC的阈值越小,Pvalue的阈值越小,筛选得到的差异表达蛋白数目越少。
步骤4.4中所述主成分分析(PCA),是一种数据分析方法。PCA通过线性变换将数据变换为一组各维度线性无关的表示,可用于提取数据的主要特征分量,常用于高维数据的降维;本发明中PCA图能够初步判定样本间差异大小,在图中两个样本之间的连线越长,则说明两个样本之间的差异越大,有利于初步判断组间样本差异和组内样本差异。
步骤五、结果导出保存
步骤5.1、按照不同的差异标准,从3个水平进行差异蛋白筛选,统计不同组别之间的差异蛋白数目,并将统计结果导出到excel表格中。
步骤5.2、根据主成分分析结果,绘制主成分图,并保存到pdf文件中。
步骤5.1中所述差异标准是指组别间的差异倍数(FC)和显著性(Pvalue)阈值的设定标准,本发明通过调节FC阈值,分别从3个不同水平去筛选组别间差异蛋白。
FC说明的是蛋白表达量变化幅度的大小,且可以分为上调FC>1,下调FC<1,无变化FC=1,上调和下调统称为有差异。Pvalue则是说明结果的可信度,一般都是越小说明结果越可靠,一般取Pvalue<0.05即可。
第1个水平:以FC>1.2同时Pvalue<0.05或者FC<0.83同时Pvalue<0.05筛选差异蛋白,并统计其数目。
第2个水平:以FC>1.5同时Pvalue<0.05或者FC<0.67同时Pvalue<0.05筛选差异蛋白,并统计其数目。
第3个水平:以FC>2.0同时Pvalue<0.05或者FC<0.5同时Pvalue<0.05筛选差异蛋白,并统计其数目。
常规操作:一般分析时会选择一个FC阈值,筛选差异蛋白,如果FC的阈值过于宽松(FC离1越近,比如0.9和1.1离1的距离是0.1,1.5和0.5离1的距离是0.5),则得到的差异蛋白太多,对后续分析不利。如果FC的阈值设置的过于严格(FC离1越远),则得到的差异蛋白过少,对后续的分析同样不利)。
为了更加直观的看到各个水平(本发明是3个水平)差异蛋白的数目,本发明同时给出了3个水平上差异蛋白的数目统计信息。
步骤5.1中所述3个水平进行差异蛋白筛选,是同时完成3个水平的差异蛋白数目统计。
步骤5.2中所述绘制主成分图,是选择第一主成分和第二主成分绘制主成分分析图。
步骤5.2中所述主成分图是一种散点图,同一组别的样本在图中的形状相同,不同组别的样本在图中的形状不同。
主成分分析图可以用来判断组内重复性和组间差异性的大小,如果一个组内的各样本之间直线距离很短,说明该组内样本的重复性很好、相似度很高。如果不同组别间各样本的直线距离很长,说明两个组别的差异性较大。如果同一组别各样本之间的直线距离很长,说明该组内样本的重复性很差;如果不同组别各样本之间的距离较短,则说明不同组别间的差异性较小。
本发明还提出了一种实现上述方法的系统,所述系统包括项目列表文件解析模块、项目解析文件准备模块、谱图自动解析启动模块、数据自动分析模块、结果导出模块;
其中,所述项目列表文件解析模块用于读取并遍历TMT项目列表文件,将符合条件的记录放入python列表中;
所述项目解析文件准备模块用于获取需要的文件路径,根据标记信息文件和定量模板文件,生成项目的定量方法文件;/>
所述谱图自动解析启动模块用于导入解析需要的数据,设定解析流程、方法、参数,启动谱图自动解析软件;
所述数据自动分析模块用于对设定好的谱图进行自动解析,获得蛋白表格、肽段表格、肽段匹配表格、蛋白信息统计表格,求出不同组别之间每个蛋白的差异倍数FC和Pvalue,并根据蛋白表达丰度的信息,按组别对样本进行主成分分析;
所述结果导出模块用于筛选组别间差异蛋白,统计不同组别之间的差异蛋白数目,绘制主成分图并保存。
本发明还提供了一种上述方法在TMT项目图谱解析和数据分析中的应用。
本发明还提供了一种TMT项目图谱解析和数据分析设备,包括:存储器和处理器;
所述存储器上存储有计算机程序,当所述计算机程序被所述处理器执行时,实现上述的方法。
本发明还提供了一种计算机可读存储介质,其上存储有计算机程序,所述计算机程序被处理器执行时,实现上述的方法。
本发明的有益效果包括:本发明可以自动定位,即自动确定相关文件在某个目录的路径,文件路径是实验人员设置的,当分析人员要用到这个路径时,可以由分析人员自己寻找,或向实验人员询问文件的路径,或用get_rawdata_path方法自动去寻找路径,程序省去了沟通环节、可以直接获取到目标位置。
谱图解析所需文件信息,自动将谱图解析所需文件添加到解析软件中,自动进行参数设置并开始解析过程,实时监督谱图解析状态,自动开启数据分析工作,减少了谱图解析和数据分析工作的人力成本,提升了项目的交付效率。通过采用本发明方法,可以把分析人员从繁杂的工作中解放出来。本发明方法不再需要人工手动去查找项目的原始数据和蛋白数据库的位置,本发明方法不需要实验人员确定标记定量方法,本方法不需要分析人员手动将谱图解析数据添加到谱图解析软件中,本方法不需要人工来监督谱图解析过程是否完成,本方法不需要分析人员手动分析相关数据,以上操作过程在本方法中均实现了自动化。本系统有效减轻的分析人员的工作量,有效的提高的项目的交付效率,显著的降低了该项工作的人力成本。
附图说明
图1a显示为本发明一实施例中TMT项目数据分析流程图。
图1b显示为本发明一实施例中TMT项目数据分析系统模块图。
图2显示为本发明一实施例中TMT项目列表文件信息图。
图3显示为本发明一实施例中TMT项目标记信息示例图。
图4显示为本发明一实施例中TMT项目蛋白数据库示例图。
图5显示为本发明一实施例中TMT项目原始数据文件图。
图6显示为本发明一实施例中定量模板文件示意图。
图7显示为本发明一实施例中定量方法文件示意图。
图8显示为本发明一实施例中软件相关按钮位置示意图。
图9显示为本发明一实施例中软件相关按钮位置示意图。/>
图10显示为本发明一实施例中软件相关按钮位置示意图。
图11显示为本发明一实施例中软件相关按钮位置示意图。
图12显示为本发明一实施例中软件相关按钮位置示意图。
图13显示为本发明一实施例中软件相关按钮位置示意图。
图14显示为本发明一实施例中软件相关按钮位置示意图。
图15显示为本发明一实施例中谱图解析结果文件示意图。
图16显示为本发明一实施例中项目信息统计表格内容示意图。
图17显示为本发明一实施例中项目分组文件内容示意图。
图18显示为本发明一实施例中项目对比文件内容示意图。
图19显示为本发明一实施例中蛋白表格文件内容示意图。
图20显示为本发明一实施例中蛋白差异计算结果示意图。
图21显示为本发明一实施例中主成分分析荷载值示意图。
图22显示为本发明一实施例中主成分分析解释率示意图。
图23显示为本发明一实施例中蛋白差异统计结果示意图。
图24显示为本发明一实施例中项目主成分分析图。
图25为本发明对比实施例中项目文件存放位置示意图。
图26为本发明对比实施例中项目目录子文件夹示意图。
图27为本发明对比实施例中生产附件文件夹示意图。
图28为本发明对比实施例中数据库文件夹示意图。
图29为本发明对比实施例中项目原始数据文件夹示意图。
图30为本发明对比实施例中定量系数添加示意图。
图31为本发明对比实施例中数据库管理界面示意图。
图32为本发明对比实施例中蛋白数据库文件窗口示意图。
图33为本发明对比实施例中谱图解析项目项目创建示意图。
图34为本发明对比实施例中图谱解析项目创建步骤弹出窗口示意图。
图35为本发明对比实施例中图谱解析进度信息示意图。
图36为本发明对比实施例中图谱解析完成后导出文件示意图。
图37为本发明对比实施例中图谱解析完成后导出文件导出条件选择示意图。
图38为本发明对比实施例中数据分析软件信息处理示意图。
具体实施方式
结合以下具体实施例和附图,对发明作进一步的详细说明。实施本发明的过程、条件、实验方法等,除以下专门提及的内容之外,均为本领域的普遍知识和公知常识,本发明没有特别限制内容。
本发明提出了一种TMT项目图谱解析和数据分析方法,目的是解决传统基于人工手动进行谱图解析和数据分析效率低下、项目交付周期长的问题。该方法包括以下步骤:项目列表文件解析,项目解析文件准备,谱图自动解析,项目数据自动分析,结果导出保存。采用本发明,可实现对多个TMT项目自动进行谱图解析和数据分析,可以避免分析人员重复劳动,节省了人力成本,提升了项目交付效率。本发明还提出了实现上述方法的系统及上述方法在TMT项目图谱解析和数据分析中的应用。
实施例一
本实施例按照项目列表文件解析、项目解析文件准备、谱图自动解析、数据自动分析和结果导出保存步骤进行。(图1a)
(1)项目列表文件解析
①读取项目列表文件(图2),利用python将文件读入内存,分析文件的每一行文字记录即每一个分析任务。
②从每一行文字记录中筛选出包含“TMT”字段,且同时包含“上机”或“搜库结题”的分析任务,并放入python列表中,本实施例中筛选得到的分析任务是“J:实验生产部蛋白实验数据2022202205MJ20220802134-TMT-6例组织样本生产附件搜库结题.txt”,其中项目的合同号是“MJ20220802134”,“搜库结题”说明本次需要进行谱图解析的是正式样本,在谱图解析完成之后需要进行数据分析。
(2)项目解析文件准备
①根据第一步中得到的TMT项目的合同号,使用get_pro_db_bjsx方法,从项目所在目录“J:实验生产部蛋白实验数据2022202208MJ20220802134-TMT-6例组织样本”中获取得到标记信息文件“J:实验生产部蛋白实验数据2022202208MJ20220802134-TMT-6例组织样本生产附件标记.xls”(图3)和蛋白数据库文件“J:实验生产部蛋白实验数据2022202208MJ20220802134-TMT-6例组织样本数据库uniprot-taxonomy_39947_unique.fasta”(图4),使用get_rawdata_path方法获取项目原始数据文件的绝对路径“K:Project MS serviceProteomics project2022Project202208MJ20220802134_LZQPP20220916222 awdata”。(图5)和谱图解析结果输出目录“K:Project MS serviceProteomics project2022Project202208MJ20220802134_LZQPP20220916222out_tmt2022_10_15_09_07_32_out_tmt”。
标记信息文件中“XE354084”是项目标记试剂的批号,“128N、129N、129C、131N、131C、132N”是项目使用的标记。
蛋白数据库文件中以“>”开头的行包含了蛋白名称和注释信息,其余行则是蛋白序列信息。
根据标记试剂批号和定量模板文件的对应关系,确定本次应用的定量模板文件是“J:hqtworkplacepyscriptsQM_templatesTMTPro 16PlexLOT_XE354084.method”(图6)使用mk_qm_method方法生成项目的定量方法文件“K:Project MS serviceProteomics project2022Project202208MJ20220802134_LZQPP20220916222out_tmt2022_10_15_09_07_32_out_tmtPro_QM.method”,该文件中128N、129N、129C、131N、131C、132N标记的name="IsActive"属性被设置为“True”(图7)
(3)谱图自动解析
①使用pd_start方法,自动启动谱图解析软件,软件完全启动后,通过get_position方法获取谱图解析软件中设置按钮(见图8中的a处)在屏幕中的坐标,通过pyautogui.leftClick点击该坐标,并进入管理界面(见图8中的b处),管理界面中有蛋白数据库管理按钮(见图8中的c处)和定量方法管理按钮(见图8中的d处)。
②使用add_db方法删除软件中已有的数据库,并添加项目数据库。add_db方法的具体操作为通过get_position方法获取谱图解析软件中蛋白数据库管理按钮在屏幕中的坐标,通过pyautogui.leftClick点击该坐标,并进入管理界面,通过get_position方法获取谱图解析软件中蛋白数据库删除按钮(见图9中的b处)在屏幕中的坐标,通过pyautogui.leftClick点击该坐标,删除软件中已有的数据库(见图9中的a处),通过get_position方法获取谱图解析软件中蛋白数据库添加按钮(见图9中的c处)在屏幕中的坐标,通过pyautogui.leftClick点击该坐标,将项目蛋白数据库文件(“J:实验生产部蛋白实验数据2022202208MJ20220802134-TMT-6例组织样本数据库uniprot-taxonomy_39947_unique.fasta”)导入软件中。
③使用add_method方法,删除谱图解析软件中已有的定量方法,导入项目对应的定量方法文件。具体操作为通过get_position方法获取谱图解析软件中定量方法管理按钮在屏幕中的坐标,通过pyautogui.leftClick点击该坐标,进入定量方法管理界面,通过get_position方法获取谱图解析软件中定量方法删除按钮(见图10中的b处)在屏幕中的坐标,通过pyautogui.leftClick点击该坐标,删除软件中已有的标记定量方法(见图10中的a处),通过get_position方法获取谱图解析软件中定量方法文件导入按钮(见图10中的c处)在屏幕中的坐标,通过pyautogui.leftClick点击该坐标,导入项目定量方法文件(“K:Project MS serviceProteomics project2022Project202208MJ20220802134_LZQPP20220916222out_tmt2022_10_15_09_07_32_out_tmtPro_QM.method”)。
④使用outdir_select方法,选择谱图解析结果保存的位置。具体操作为通过get_position方法获取谱图解析软件中新建项目按钮(见图11中的a处)在屏幕中的坐标,通过pyautogui.leftClick点击该坐标,打开新建项目界面,通过get_position方法获取谱图解析软件中谱图解析结果输出文件夹选择按钮(见图11中的b处)在屏幕中的坐标,通过pyautogui.leftClick点击该坐标,通过pywinauto.keyboard.send_key将谱图解析结果输出目录的绝对路径(“K:Project MS service
Proteomics project2022Project202208MJ20220802134_LZQPP20220916222out_tmt2022_10_15_09_07_32_out_tmt”)输入软件的文本框内,完成谱图解析结果输出目录的选择。
⑤使用workflow_select方法,选择项目的工作流程(包括processingworkflow和consensus workflow)。具体操作为通过get_position方法获取处理工作流(processing workflow)选择按钮(见图11中的c处)在屏幕中的坐标,通过pyautogui.leftClick点击该坐标,打开处理工作流选择窗口,通过pywinauto.keyboard.send_key将处理工作流的绝对路径输入文本框,完成处理工作流的选择。通过get_position方法获取共识工作流(consensus workflow)选择按钮(见图11中的d处)在屏幕中的坐标,通过pyautogui.leftClick点击该坐标,打开共识工作流选择窗口,通过pywinauto.keyboard.send_key将共识工作流的绝对路径输入文本框,完成共识工作流的选择。
⑥使用qm_select方法,选择项目的定量方法。具体操作为通过get_position方法获取软件中定量方法选择按钮(见图11中的e处)在屏幕中的坐标,并通过pyautogui.leftClick点击该坐标,完成定量方法的设置。
⑦使用rawdata_import方法将项目原始数据据文件导入谱图解析软件。具体操作为通过get_position方法获取软件中原始数据导入按钮(见图11中的f处)在屏幕中的坐标,并通过pyautogui.leftClick点击该坐标,打开原始数据选择窗口,pywinauto.keyboard.send_key将项目原始数据的绝对路径(“K:Project MS serviceProteomics project2022Project202208MJ20220802134_LZQPP20220916222 awdata”)输入窗口的文本框中,完成原始数据的导入。
⑧使用parameter_set方法,对关键参数进行设定,并开启谱图解析过程。具体操作为通过get_position方法获取软件中参数修改按钮(Edit,见图12中的a处)在屏幕中的坐标,并通过pyautogui.leftClick点击该坐标,进入工作流(workflow)设置界面,通过get_position方法获取谱图文件重校正按钮(Spectrum Files RC,见图13中的a处)在屏幕中的坐标,并通过pyautogui.leftClick点击该坐标,打开谱图文件重校正参数设置界面,通过get_position方法获取谱图解析软件中蛋白数据库选择按钮(见图13中的b处)在屏幕中的位置,通过pyautogui.leftClick点击该坐标,完成蛋白数据库的设置。通过get_position方法获取谱图解析引擎(Sequest HT,见图13中的c处)在屏幕中的坐标,通过pyautogui.leftClick点击该坐标,打开谱图解析引擎参数设置界面,通过get_position方法获取蛋白数据库选择按钮在屏幕中的位置,通过pyautogui.leftClick点击该坐标,完成蛋白数据库的设置。通过get_position方法获取执行分析按钮(Run,见图13中的d处)在屏幕中的坐标,通过pyautogui.leftClick点击该坐标。通过get_position方法获取分析警告窗口中忽略按钮(Ignore,见图14中的a处)在屏幕中的位置,通过pyautogui.leftClick点击该坐标,开启谱图解析过程。
(4)数据自动分析
①使用stat_check方法,每隔2分钟,对谱图解析结果输出目录(“K:ProjectMS serviceProteomics project2022Project202208MJ20220802134_LZQPP20220916222out_tmt2022_10_15_09_07_32_out_tmt”)中所有文件进行一次扫描,直到结果输出目录中出现名称为“Protein_information.xls”的文件(当输出目录中已经输出有包括蛋白表格(protein.xls)、肽段表格(peptide.xls)、肽段匹配表格(psm.xls)在内的文件,生成“Protein_information.xls”文件),说明谱图解析过程已经完成,且已通过TMT_export方法生成谱图解析结果(图15)。本次谱图解析生成的结果有蛋白表格(protein.xls)、肽段表格(peptide.xls)、肽段匹配表格(psm.xls)、信息统计表格(Protein_information.xls),从Protein_information.xls文件中可知,本次项目原始数据中谱图数(Total Spectrum)是179601,在蛋白数据库中有匹配的谱图数(IdentifiedSpectrum)是44659,本次从项目原始数据中鉴定到的肽段数(Peptide number)是27917,本次谱图解析共鉴定到的蛋白数(Protein number)是25949,本次谱图解析鉴定到的蛋白质组数(25949)是6419。(图16)
②利用python读取项目分组文件(图17)和对比文件(图18)、谱图解析产生的“protein.xls”文件(图19)。分组文件的第一列(sample)是本次数据分析的样本名称,第二列(group)是第一列样本的分组。对比文件中第一列(control)是对照组,第二列(other)是对比组。Protein.xls文件包含了本次谱图解析所得到的蛋白基本信息,包括蛋白的序列号(Accession)、描述信息(Description)、分子量(MW [kDa])、等电点(calc. pI)、匹配得分(Score Sequest HT: Sequest HT)、蛋白肽段数(# Peptides)和丰度信息(Abundance)。/>
③利用numpy和scipy对每一个蛋白按组别进行费歇尔精确检验,求出不同组别之间每个蛋白的差异倍数FC和Pvalue。(图20)以蛋白“E9Q616”为例,该蛋白在对照组(CK)的三个样本(young_1,young_2,young_3)中的丰度分别为0.984、0.959、1.02,在对照组中该蛋白丰度的平均值为0.987666666666666,该蛋白在对比组(CHU)中三个样本(ageing_1、ageing_2、ageing_3)中的丰度分别为1、1.062、1.086,该蛋白在该组中平均表达丰度为1.04933333333333。该蛋白在对比组和对照组中的差异倍数(FC(ageing/young))为1.062436719541,显著性(Pvalue(ageing/young))为0.118786543000299。
④利用sklearn和protein.xls文件中蛋白表达丰度的信息,进行主成分分析(PCA),并求出每一个蛋白在第一主成分和第二主成分上的荷载值(蛋白与成分之间的相关性系数,图21)和成分解释度(对变量的解释程度,图22)。以蛋白“E9Q616”为例,其在第一主成分(p1)上的荷载值为0.00683669731542218,其在第二主成分(p2)上的荷载值为0.0137218748410895。第一主成分对变量的解释率为0.724,第一主成分对变量的解释率为0.139,两个成分对变量的累计解释率达到了0.863。
(5)结果导出保存
①按照不同的差异标准,从3个水平进行差异蛋白筛选,统计不同组别之间的差异蛋白数目,并将统计结果导出到excel表格中。(图23)
第一个水平:按照上调倍数FC>1.2且Pvalue<0.05筛选得到表达蛋白225个, 按照下调倍数FC<0.83且Pvalue<0.05筛选得到表达蛋白数目为922个,该水平差异蛋白总数为1147个。
第二个水平:按照上调倍数FC>1.5且Pvalue<0.05筛选得到表达蛋白71个, 按照下调倍数FC<0.67且Pvalue<0.05筛选得到表达蛋白数目为272个,该水平差异蛋白总数为343个。
第三个水平:按照上调倍数FC>2.0且Pvalue<0.05筛选得到表达蛋白22个, 按照下调倍数FC<0.5且Pvalue<0.05筛选得到表达蛋白数目为44个,该水平差异蛋白总数为66个。
②根据主成分分析结果,按组对样本绘制主成分散点图,并保存到pdf文件中。(图24)图中横轴代表的是第一主成分(PC1),纵轴代表的是第二主成分(PC2),图中圆点代表的是对比组(CHU)中的三个样本(ageing_1、ageing_2、ageing_1),三角形代表的是对照组(CK)中的三个样本(young_1、young_3、young_3),可以看到两个组别被第一主成分分成两部分,说明两组样本之间存在较为明显的差异,同时说明第一主成分可以对两个组别起到很好的区分效果。
对比实施例
本示例是以往常规的、人工手动处理的方法。
(1)项目列表文件解析
以往该过程需要分析人员先找到项目列表文件,并通过记事本或者WPS打开该文件,由于文件有多行,每一行代表了不同类型项目(TMT、label free、DIA等等)的不同分析需求。分析人员需要从多行记录中筛选(根据字段“TMT”字段,且同时包含“搜库结题”)出需要进行谱图解析的TMT项目信息(比如J:实验生产部蛋白实验数据2022202205MJ20220509107-TMT-6例组织样本生产附件搜库结题.txt),主要是合同号(MJ20220509107,其他信息程序不使用)。
(2)项目解析文件准备
根据第一步得到的合同号MJ20220509107找到该项目相关文件的存放位置(某电脑的某盘符的某个文件夹的某个子文件夹的某个子文件夹下面,图25),如果某个文件夹下面文件比较多可能需要仔细用眼睛去寻找或者检索合同号,如果文件放错位置即本来应该放到A文件夹下却错误的放到B文件夹下面,又会浪费分析人员不少时间。该项目的路径要么分析人员自己手动寻找,要么通过邮件或者钉钉等方式向生产人员或者运营人员索要。
在通过各种方式得到该目录(J:实验生产部蛋白实验数据2022202205MJ20220509107-罗朝兵-罗朝兵-TMT-6例组织样本)后,打开该目录,观察其中的子文件夹(图26),标记文件是放在“生产附件”的文件夹中(图27),蛋白数据库是放在“数据库”文件夹中(J:实验生产部蛋白实验数据2022202205MJ20220509107-罗朝兵-罗朝兵-TMT-6例组织样本数据库uniprot-taxonomy_39947_unique.fasta,图28),项目的原始数据是放在另一个电脑的某一个文件夹下(K:Project MS serviceProteomics project2022Project202208MJ20220802134_LZQPP20220916222 awdata,图29,这个文件夹不是由分析人员创建,里面的数据也不是由分析人员放进去,分析人员只是知道在那个电脑的某个盘符下,试着去找,不确定一定能够找到,也不确定里面有没有放数据)。找到这几个数据的路径之后,要么打开2个文件夹窗口,要么一次打开这3个文件夹,要么把这3个文件夹的路径记录在记事本和其他工具上,以便后续添加数据。
通过标记信息文件确定标记的批号,然后根据该批号的说明书添加定量系数(图30),然后根据项目标记试剂使用的试剂情况在软件中逐一输入标记和对应的定量文件,不同项目使用的标记个数不一定是16个,也可能是10个,剩下6个没使用,而且不同项目使用的标记不一定有严格的规律,这就给手动制作项目的定量方法文件造成了很多麻烦,也使得分析人员也有一定的抵触情绪。
(3)手动操作软件进行谱图解析
在以上文件都准备完整后,就可以开始谱图解析工作。通过常规的点击桌面图标,启动PD软件(和在电脑上打开QQ是一个道理),并等待软件完全打开(一些桌面软件在打开时都有加载的过程,没有加载完成时是无法使用软件功能的,软件是否加载完全是通过人眼观察来判断),后面的操作可以参考自动化操作,因为软件的使用教程都是一样的,无外乎是人还是机器去点击软件上那些按钮。
本对比实施例中会尽量把所有人工操作和机器操作不同的地方都进行详细的说明。
在进软件后还是通过数据库管理按钮(见图31中的a处)进入数据库管理界面,通过蛋白数据库添加按钮(见图31中的b处)添加蛋白数据库,当点击数据库添加按钮之后,电脑会弹出来一个用来选择蛋白数据库文件窗口(图32),通过手动调整的方式找到蛋白数据库(J:实验生产部蛋白实验数据2022202205MJ20220509107-罗朝兵-罗朝兵-TMT-6例组织样本数据库uniprot-taxonomy_39947_unique.fasta)并添加到软件中。
接下来开始创建谱图解析项目,通过手动点击软件中的项目创建按钮(见图33中的a处),软件弹出一个窗口(图34),可以通过按钮a来设置谱图解析结果输出目录,通过按钮b和按钮c来设置项目使用的工作流程,通过按钮d来设置定量方法文件,通过按钮e来添加原始数据,这些和自动化差不多,只是通过人手动点击罢了。
剩下的步骤可以参考实施例中图36至图38这些步骤都是大同小异,只是手动依次点击罢了。
谱图解析过程开启之后,就是等待谱图解析过程完成,不同的项目谱图解析消耗时间不同,如果原始数据的文件大小和蛋白数据库的文件大小较大,则耗时较久,如果电脑负载较高则可能消耗的时间也较久,也就是说谱图解析到底要耗费都长时间是没有办法确定的,只能通过分析人员不断地去观察软件中谱图解析的进度信息(见图35中的a处),如果进度为100%(见图35中的b处)则说明解析过程已完成,否则说明解析过程未完成(见图35中的c处),仍需等待谱图解析完成,方可进行下一步的数据分析工作。
在分析人员观察到谱图解析过程完成之后,通过点击软件中的“文件按钮”(见图36中的a处)和“导出按钮”(见图36中的b处)和“导出Excel按钮”(见图36中的c处),弹出窗口(图37),依次导出“proteins”、“Protein Groups”、“Peptide Groups”、“PSMs”和“MS/MS Spectrum Info”表格,并对以上表格进行处理,从中整理出“protein_infomation.xls”表格。
(4)手动进行数据分析
手动数据分析内容与自动进行数据分析基本一样,不同之处在于操作过程不同。手动进行数据分析同样需要protein表格,分组对比文件(参考图17-图19),只不过自动化是把文件通过python读入内存,不需要把文件打开,而手动需要用wps把分组对比文件打开。
然后通过python自研数据分析软件(图38)对这些信息进行处理,将谱图解析结果路径输入"谱图解析结果文件夹路径"对应的文本框中(见图38中的a处),将“protein_infomation.xls”的路径输入“蛋白信息表”对应的文本框中(见图38中的b处),将蛋白数据库的路径输入“数据库文件路径”对应的文本框(见图38中的c处),将样本信息输入“样本”对应的文本框(见图38中的d处),将分组信息输入“分组”对应的文本框(见图38中的e处),将对照组信息输入“对照组”对应的文本框(见图38中的f处),将对比组信息输入“对比组”对应的文本框中(见图38中的g处),然后点击确认按钮即可开始数据分析工作,数据分析结果与自动化分析结果基本一致。本部分使用的数据分析软件已经整合到自动化数据分析流程中,且不需要信息输入弹窗(图38)。
本发明的保护内容不局限于以上实施例。在不背离发明构思的精神和范围下,本领域技术人员能够想到的变化和优点都被包括在本发明中,并且以所附的权利要求书为保护范围。/>
- 一种模型应用方法和数据分析系统
- 一种基于大数据分析的变电设备非项目类成本预测方法
- 一种数据分析装置、数据分析处理系统和数据分析方法
- 数据分析装置、数据分析处理系统和数据分析方法