化合物N1-(3-氨丙基)-N3-环己基-1,3-丙二胺及其制备方法和应用
文献发布时间:2023-06-19 19:30:30
技术领域
本发明涉及一种新化合物N
背景技术
目前应用于环氧固化剂领域的胺主要有三类:脂环胺、脂肪胺和芳香胺,通常的脂环胺(例如HMDA、IPDA和1,3-BAC)具有制品耐黄变能力强、制品硬度高等特点,但由于分子的脂肪链不够长,导致其制品冲击强度较差;脂肪胺虽然其冲击强度较好,但分子的挥发性强,毒性较大,制品的硬度较差;芳香胺由于其毒性较大以及耐黄变能力差,目前逐渐被禁止使用。
随着环氧固化剂下游应用的迅猛发展,对具有特定结构的新分子胺有了一定的需求,需要开发一款可以使制品兼顾冲击强度和硬度的环氧固化剂,以满足下游客户的需求。
发明内容
基于现有技术中存在的上述不足,本发明提供了一种新化合物N
同时,本发明还提供了上述化合物作为环氧树脂固化剂的应用,得到的制品不但满足兼顾冲击强度和硬度的需求,同时还具有耐黄变能力强、玻璃化转化温度高等优点。
为了实现上述发明目的,本发明采用如下的技术方案:
本发明提供一种化合物N
本发明还提供一种上述式(1)所示N
1)将N-(3-氨基丙基)环己胺、水和助剂混合,然后加入丙烯腈进行反应,反应结束后再经减压精馏得到中间体;
2)将雷尼催化剂、碱性改性剂和溶剂混合,之后通入氢气调节体系至反应压力,并加热至反应温度,然后在氢气氛围下加入步骤1)制得的中间体,进行加氢反应,制得式(1)所示N
本发明的方法涉及的主要反应式如下:
本发明中,步骤1)所述丙烯腈与N-(3-氨基丙基)环己胺的摩尔比为0.8-0.99:1,优选0.9-0.95:1;通过控制丙烯腈与氨丙基环己胺的摩尔比低于1:1可以有效的减少双腈的生成。
本发明中,步骤1)所述水与N-(3-氨基丙基)环己胺的摩尔比为0.25-2:1,优选0.5-1:1。
本发明中,步骤1)所述助剂选自N,N-二甲基乙醇胺和/或N,N-二乙基乙醇胺;
所述助剂的用量为N-(3-氨基丙基)环己胺质量的5-20wt%,优选8-15wt%。
本发明中,步骤1)得到的所述中间体为氰乙基氨丙基环己胺,具有下式(2)所示的结构:
根据式(1)化合物的结构特点,可以采用先通过N-(3-氨基丙基)环己胺与丙烯腈反应得到上式(2)的中间体(氰乙基氨丙基环己胺),再通过加氢即可得到目标产品。但本发明人在研究中发现,氨丙基环己胺与丙烯腈反应的过程中会生成两种双腈类化合物,结构如下:
而双腈1和双腈2在加氢的过程中,极易发生分子内成环和分子间脱氨而形成大分子副产物,大分子副产物对催化剂表面孔道的堵塞、活性位点的覆盖会导致催化剂快速失活。为此,本发明研发人员通过进一步研究发现,在反应体系中引入助剂N,N-二甲基乙醇胺和/或N,N-二乙基乙醇胺,可以有效解决上述问题。本发明采用的N,N-二甲基乙醇胺和/或N,N-二乙基乙醇胺其结构中同时含有特定位置的羟基和叔胺基团,一方面,其中的羟基可以与中间体上的仲氮形成氢键,从而产生一定的空间位阻,阻止中间体进一步的与丙烯腈反应得到双腈1和双腈2,从而降低这两种副产在下一步加氢过程中对催化剂毒害;另一方面,叔胺具有很强的空间位阻,在本发明的条件下不易与丙烯腈发生反应,避免生成新的杂质。此外,本发明的助剂沸点较低,可通过简单的精馏实现原料的回用。
本发明中,步骤1)所述反应,温度为10-40℃,优选20-30℃;保温反应时间为0.2-2h,优选0.5-1h。
本发明中,步骤1)所述丙烯腈采用连续加料方式,优选滴加加料,加料时间为2-6h,优选3-4h;加料结束后继续保温反应,加料时间不计入上述保温反应时间内。
本发明中,步骤1)所述减压精馏,真空度为0.5-5KPa,优选1-2KPa,温度为88-150℃,优选100-112℃;通过减压精馏可以脱除反应液中的N-(3-氨基丙基)环己胺、水和助剂,脱除的N-(3-氨基丙基)环己胺、水和助剂可以回收使用,可以为混合物或纯净物,优选混合物,以降低能耗和操作复杂性。
本发明中,步骤2)所述雷尼催化剂选自雷尼镍、雷尼钴中的一种或多种,优选Grace2400、Grace2800、Grace 2724、迅凯1200、迅凯3300中的一种或多种;
所述雷尼催化剂的用量为中间体质量的10-30wt%,优选15-25wt%。
本发明中,步骤2)所述碱性改性剂为碱性化合物,选自氢氧化锂、氢氧化钠、氢氧化钾、氨水(如浓度为25wt%的氨水),优选氢氧化锂、氢氧化钠中的一种或多种;优选地所述碱性改性剂使用前配制为浓度5-15wt%的水溶液;
所述碱性改性剂的用量为雷尼催化剂质量的2-20wt%,优选5-10wt%。
本发明中,步骤2)所述溶剂选自甲醇、乙醇、四氢呋喃、二氧六环中的一种或多种,优选乙醇和/或四氢呋喃;
所述溶剂的用量为中间体质量的0.5-2倍,优选0.75-1.25倍。
本发明中,步骤2)反应压力为3-8MPaG,优选4-6MPaG;所述反应温度为70-140℃,优选80-120℃。
本发明中,步骤2)所述中间体采用连续加料方式,优选滴加加料,加料时间为2-8h,优选4-6h;加料结束后继续保温进行加氢反应。
本发明中,步骤2)所述加氢反应,在加料结束后的反应时间为0.2-2h,优选0.4-0.6h(上述中间体加料时间不计入反应时间)。
本发明中,步骤2)在加氢反应结束后,还包括降温、过滤等操作得到包含N
同时,本发明还提供了上述式(1)所示N
与现有技术相比,本发明技术方案有益效果在于:
(1)本发明研发出一种N
(2)本发明工艺通过引入的助剂,可以显著降低双腈1和双腈2的含量,使双腈总含量≤0.5%,中间体的选择性≥99%,双腈杂质降低至此含量后,可以消除后续加氢反应中对于催化剂的毒害,催化剂性能稳定,可连续套用30批次以上,产品收率≥98%。
(3)本发明未反应的N-(3-氨基丙基)环己胺、助剂和水可以实现回用,助剂无需再次添加,可以有效的降低物耗以及三废的产生,是一条经济、绿色、可持续发展的工艺,符合国家的发展战略。
附图说明
图1为实施例1中由步骤1)制备的中间体的质谱图;
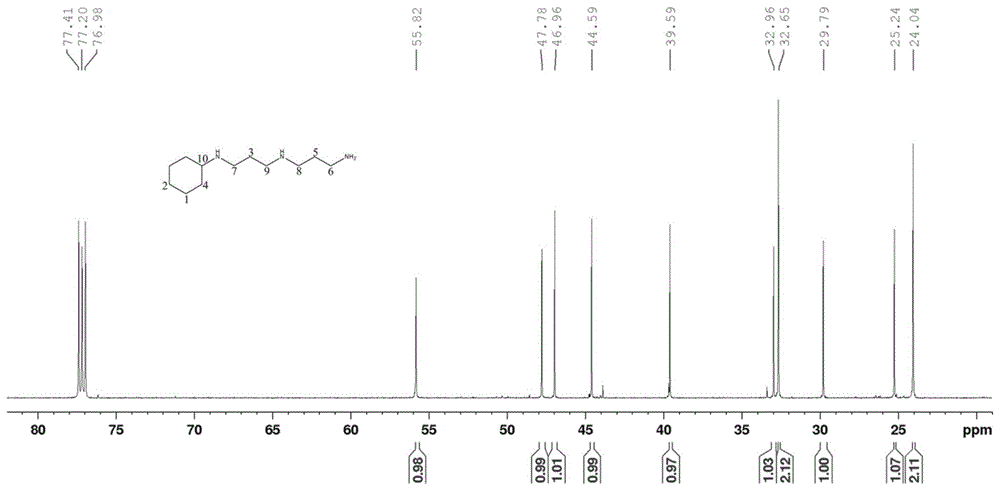
图2为实施例1中由步骤2)制备的产品的核磁碳谱图。
具体实施方式
为了更好的理解本发明的技术方案,下面的实施例将对本发明所提供的方法予以进一步的说明,但本发明不限于所列出的实施例,还应包括在本发明的权利要求范围内其他任何公知的改变。
以下实施例中进行气相色谱分析的条件为:安捷伦DB-5色谱柱,进样口温度280℃,FID检测器温度300℃,柱流速1.3ml/min,氢气流速40ml/min,空气流速400ml/min,程序升温方式为:50℃保持2min,以5℃/min升温至80℃,然后以15℃/min升温至300℃,保持15min。
以下实施例中进行GC-MS分析条件为:安捷伦7890A GC System-5975CMSD,DB-5MS色谱柱,进样口温度280℃,进样量0.2μL,分流比30,柱温为50℃保持2min,以5℃/min升至80℃,再以15℃/min升至280℃,保持5min;连接线温度300℃,检测器为四级杆质谱,离子源温度为230℃,四级杆温度为200℃,质量数扫描范围为29-800。
以下实施例中进行核磁碳谱分析条件为:布鲁克AVANCE NEO600MHz,13C频率:150.93MHz,脉冲序列:zgig30,溶剂:CDCl3,采样点数TD:64K,脉冲前恢复时间D1:3s,扫描次数NS:1024。
以下实施例或对比例中所用的主要原料来源如下,其它若未特别说明,均为普通市售原料:
N-(3-氨基丙基)环己胺:工业级,购于索尔维精细化工有限公司;
雷尼镍催化剂:Grace 2400/2800,购于Grace催化剂公司;迅凯1200,购于上海迅凯新材料科技有限公司。
雷尼钴催化剂:Grace 2724,购于Grace催化剂公司;迅凯3300,购于上海迅凯新材料科技有限公司。
主要设备信息如下:
高压反应釜:规格1L,制造商为烟台科立设备;
平流泵:型号2PB00C,制造商为北京卫星。
实施例1
制备化合物N
1)将156g(1mol)N-(3-氨基丙基)环己胺、18g(1mol)水、15.6gN,N-二甲基乙醇胺加入到反应釜中,升温至30℃,开始往反应釜中滴加丙烯腈,滴加时间为4h,丙烯腈的滴加量为47.7g(0.9mol),再在30℃下保温0.5h停止反应,得到中间体反应液,中间体即氰乙基氨丙基环己胺,采用质谱表征,结果如图1所示。取样进行气相色谱分析,丙烯腈转化率100%,中间体选择性99.5%,双腈1的选择性为0.12%,双腈2的选择性为0.16%。
将反应液进行减压精馏,在真空度为3KPa,将塔顶温度在≤126℃的馏分进行收集,经色谱分析,全部为N-(3-氨基丙基)环己胺、水和N,N-二甲基乙醇胺,可以用于下次回用,塔底得到中间体(其中双腈1和双腈2总含量为0.28wt%),用于下步加氢反应。
2)在高压反应釜中加入10g雷尼镍催化剂(Grace 2800),0.5g氢氧化钾(溶于水形成10wt%水溶液)和100g乙醇,用氮气和氢气依次置换3遍后,充氢至2MPaG,开始升温,当反应温度升至90℃时,将氢气压力充压至5MPaG并且维持此压力,用平流泵往反应体系中匀速滴加中间体,当进料至50g时,总共滴加时间为3h,停止进料,继续反应0.4h,降温、过滤得到包含N
重复上述步骤制备化合物N
表1
/>
实施例2
制备化合物N
1)将156g(1mol)N-(3-氨基丙基)环己胺、9g(0.5mol)水、23.4gN,N-二乙基乙醇胺加入到反应釜中,升温至40℃,开始往反应釜中滴加丙烯腈,滴加时间为2h,丙烯腈的滴加量为42.4g(0.8mol),再在40℃下保温2h停止反应,得到中间体反应液,取样进行气相色谱分析,丙烯腈转化率100%,中间体选择性99.62%,双腈1的选择性为0.08%,双腈2的选择性为0.13%。
将反应液进行减压精馏,在真空度为1KPa,将塔顶温度在≤100℃的馏分进行收集,经色谱分析,全部为N-(3-氨基丙基)环己胺、水和N,N-二乙基乙醇胺,可以用于下次回用,塔底得到中间体(其中双腈1和双腈2总含量为0.21wt%),用于下步加氢反应。
2)在高压反应釜中加入7.5g雷尼钴催化剂(Grace 2724),0.15g氢氧化锂(溶于水形成10wt%水溶液)和62.5g四氢呋喃,用氮气和氢气依次置换3遍后,充氢至1MPaG,开始升温,当反应温度升至140℃时,将氢气压力充压至3MPaG并且维持此压力,用平流泵往反应体系中匀速滴加中间体,当进料至50g时,总共滴加时间为2h,停止进料,继续反应0.6h,降温、过滤得到包含N
重复上述步骤制备化合物N
表2
/>
实施例3
制备化合物N
1)将156g(1mol)N-(3-氨基丙基)环己胺、36g(2mol)水、7.8gN,N-二甲基乙醇胺加入到反应釜中,升温至20℃,开始往反应釜中滴加丙烯腈,滴加时间为3h,丙烯腈的滴加量为50.35g(0.95mol),再在20℃下保温1h停止反应,得到中间体反应液,取样进行气相色谱分析,丙烯腈转化率100%,中间体选择性99.45%,双腈1的选择性为0.18%,双腈2的选择性为0.20%。
将反应液进行减压精馏,在真空度为0.5KPa,将塔顶温度在≤88℃的馏分进行收集,经色谱分析,全部为N-(3-氨基丙基)环己胺、水和N,N-二甲基乙醇胺,可以用于下次回用,塔底得到中间体(其中双腈1和双腈2总含量为0.38wt%),用于下步加氢反应。
2)在高压反应釜中加入15g雷尼镍催化剂(迅凯1200),1.5g氢氧化钠(溶于水形成10wt%水溶液)和37.5g甲醇,用氮气和氢气依次置换3遍后,充氢至2MPaG,开始升温,当反应温度升至80℃时,将氢气压力充压至4MPaG并且维持此压力,用平流泵往反应体系中匀速滴加中间体,当进料至50g时,总共滴加时间为4h,停止进料,继续反应0.2h,降温、过滤得到包含N
重复上述步骤制备化合物N
表3
/>
实施例4
制备化合物N
1)将156g(1mol)N-(3-氨基丙基)环己胺、4.5g(0.25mol)水、31.2gN,N-二乙基乙醇胺加入到反应釜中,升温至10℃,开始往反应釜中滴加丙烯腈,滴加时间为6h,丙烯腈的滴加量为52.47g(0.99mol),再在10℃下保温0.2h停止反应,得到中间体反应液,取样进行气相色谱分析,丙烯腈转化率100%,中间体选择性99.21%,双腈1的选择性为0.24%,双腈2的选择性为0.23%。
将反应液进行减压精馏,在真空度为2KPa,将塔顶温度在≤112℃的馏分进行收集,经色谱分析,全部为N-(3-氨基丙基)环己胺、水和N,N-二乙基乙醇胺,可以用于下次回用,塔底得到中间体(其中双腈1和双腈2总含量为0.47wt%),用于下步加氢反应。
2)在高压反应釜中加入5g雷尼钴催化剂(迅凯3300),1g氨水(质量浓度为25%)和50g二氧六环,用氮气和氢气依次置换3遍后,充氢至4MPaG,开始升温,当反应温度升至120℃时,将氢气压力充压至8MPaG并且维持此压力,用平流泵往反应体系中匀速滴加中间体,当进料至50g时,总共滴加时间为6h,停止进料,继续反应0.5h,降温、过滤得到包含N
重复上述步骤制备化合物N
表4
/>
实施例5
制备化合物N
将实施例1反应液进行减压精馏,在真空度为1KPa,将塔顶温度为125℃的馏分进行收集,经色谱分析,丙烯腈转化率100%,中间体的纯度为99.85%(其中双腈1和双腈2总含量为0.04wt%),用于下步加氢反应。
2)在高压反应釜中加入12.5g雷尼镍催化剂(Grace 2400),1.25g氢氧化钠(溶于水形成10wt%水溶液)和25g乙醇,用氮气和氢气依次置换3遍后,充氢至3MPaG,开始升温,当反应温度升至70℃时,将氢气压力充压至6MPaG并且维持此压力,用平流泵往反应体系中匀速滴加中间体,当进料至50g时,总共滴加时间为4h,停止进料,继续反应2h,降温、过滤得到包含N
对比例1
参照实施例1中方法,不同之处仅在于:步骤1)中不加入N,N-二甲基乙醇胺,其它参数和操作不变,制得中间体,取样进行气相色谱分析:丙烯腈转化率100%,中间体选择性97.82%,双腈1的选择性为1.08%,双腈2的选择性为0.94%。
将上述中间体按照实施例1步骤2)方法用于加氢反应,取样进行气相色谱分析,经分析中间体的转化率为95.78%,N
对比例2
参照实施例1中方法,不同之处仅在于:步骤2)中不加入碱性改性剂氢氧化钾,其它参数和操作不变,制得包含N
本发明制备的化合物N
将N
将上述制得的树脂产品,按照下述方法测试其各项性能,结果如表5所示:
冲击强度:参照《GB/T 1843-2008塑料悬臂梁冲击强度的测定》;
硬度:制品固化24h,参照《GB/T 2411-1980塑料邵氏硬度试验方法》,HTS-800D,上海奕纵精密仪器有限公司;
耐黄变:采用QUV试验机,于辐照度0.35w/m2@340nm下处理24h,测试色差变化ΔE;
玻璃化转化温度(Tg):DSC测试,10-150℃,升温速率,5℃/min。
表5
尽管本发明的内容已经通过上述优选实施例作了详细介绍,但应当认识到上述的描述不应被认为是对本发明的限制。本领域技术人员可以理解,在本说明书的教导之下,可对本发明做出一些修改或调整。这些修改或调整也应当在本发明权利要求所限定的范围之内。
- 一种N-(3-氨基丙基)-1,3-丙二胺的制备方法
- 一种N-(3-氨基丙基)-1,3-丙二胺的制备方法