一种检测2,6-二氧杂螺4,5癸烷类化合物或其盐中杂质的方法
文献发布时间:2023-06-19 19:30:30
技术领域
本发明属于药物分析领域,具体涉及采用高效液相色谱方法检测2,6-二氧杂螺[4,5]癸烷类化合物或其盐中杂质的方法。
背景技术
阿片受体是一类以阿片样肽为配体的G蛋白偶联受体,μ、κ和δ受体是经典的三类阿片受体。阿片受体在体内分布广泛,但在神经系统的分布不均匀。在脑内、丘脑内侧、脑室及导水管周围灰质阿片受体密度较高,这些结构与痛觉的整合及感受有关。阿片类药物是目前临床上最有效的镇痛药物,但通常容易引起一些与靶点相关的副作用,如:呼吸抑制,便秘等。阿片类GPCR受体与配体结合后可以同时影响多条下游信号通路,包括Gi蛋白信号通路及β-Arrestin信号通路。目前研究表明,阿片类药物的镇痛作用来源于μ受体的Gi蛋白信号通路,而相关的副作用,如:呼吸抑制,便秘等,则与μ受体下游的β-Arrestin信号通路相关。与野生型小鼠相比,在β-Arrestin-2基因敲除的小鼠上注射吗啡后镇痛作用增强,且药效时间延长,而相关不良反应减弱。因此,Gi蛋白偏向性μ受体激动剂可以选择性激活Gi信号通路,而对β-Arrestin通路没有影响,或影响较小。因此,可以预期Gi蛋白偏向性μ受体激动剂在临床上有更好的镇痛作用,而阿片类相关不良反应则大大减少。
中国发明专利CN111148515 B公开了一系列Gi蛋白偏向性μ受体激动剂,其中包括式(I)所示的2,6-二氧杂螺[4,5]癸烷类化合物。
按照CN111148515B公开的制备方法得到的式(I)化合物含有多种杂质,且结构类似,因此大大增加了同时检测原料药中多种杂质的难度。而药物中含有杂质会降低其疗效和影响其稳定性,甚至对人体健康有害或产生其他副作用。因此,对杂质进行检测,控制药物纯度,以控制药物质量至关重要。
为了有效控制式(I)化合物及其盐的杂质含量,提高药物安全性,有必要开发一种专属性好、分离度高的检测方法。
发明内容
本发明提供了一种检测式(I)化合物或其盐中杂质的方法。
本发明采用的技术方案如下。
采用高效液相色谱技术,色谱柱以十八烷基硅烷键合硅胶为填充剂,流动相A为辛烷磺酸钠及磷酸二氢钾的缓冲盐溶液,流动相B为乙腈。
在一些实施方式中,所述杂质包括杂质A、B、C、D、E、F、G和/或H,其结构式如下所示:
在一些实施方式中,上述任意一种检测方法所述的色谱柱为Waters SymmetryShield RP18或Inertsil ODS-2,优选为Waters Symmetry Shield RP18。
在一些实施方式中,上述任意一种检测方法所述的流动相A中还含有三乙胺。
在一些实施方式中,上述任意一种检测方法所述的流动相A中含有0.0025 mol/L辛烷磺酸、0.01 mol/L磷酸二氢钾、0.1%三乙胺。
在一些实施方式中,上述任意一种检测方法所述的流动相A的pH为2.5 ± 0.05,优选以磷酸调节pH。
在一些实施方式中,上述任意一种检测方法所述的流动相采用等度洗脱,流动相A与流动相B比例为76:24。
在一些实施方式中,上述任意一种检测方法所述的流动相采用梯度洗脱,梯度条件如下:
在一些实施方式中,上述任意一种检测方法所述的流动相A的制备方法包括:称取辛烷磺酸钠、磷酸二氢溶于水中,加入三乙胺,用磷酸调节pH至2.5±0.05,混匀,超声过滤脱气,即得。
在一些实施方式中,上述任意一种检测方法所述的流动相流速为0.9 ~ 1.1 mL/min,优选为1.0 mL/min。
在一些实施方式中,上述任意一种检测方法所述的色谱柱的柱温为33 ~ 37 ℃,优选为35 ℃。
在一些实施方式中,上述任意一种检测方法所述的盐为马来酸盐、盐酸盐、硫酸盐、醋酸盐、酒石酸盐、磷酸盐、硫酸盐、苹果酸盐、富马酸盐或柠檬酸盐。所述盐可采用本领域技术人员常规的方法进行制备,例如在纯的溶液或合适的惰性溶剂中用足够量的酸与式(I)化合物接触的方式获得酸加成盐。
在本发明的一个方面中,所述检测方法,包括如下步骤:
(1)制备稀释剂。
(2)制备供试品溶液:称定式(I)化合物或其盐,用稀释剂进行溶解,配制成供试品溶液。
在一些实施方式中,上述任意一种检测方法中,所述稀释剂含有乙腈和水,乙腈与水的体积比为1:1 ~ 1:4。
在一些实施方式中,上述任意一种检测方法中,采用面积归一化法或外标法计算杂质含量。
杂质A~G可按照常规方法进行制备,例如杂质A~E和杂质G可参照CN111148515B的制备方法进行制备,杂质H可采取以下方法制备:
。
本发明具有如下的优点及有益效果:
1. 具有优异的专属性,可以对多种结构类似的杂质实现有效分离;主成分与相邻杂质、各杂质之间的分离度均大于1.5。
2. 能同时检测多种杂质,效率更高,成本更低。
3. 灵敏度高,检测限和定量限低,能够对痕量的的杂质进行检测和定量分析。
附图说明
图1为实施例2中三氟乙酸体系下色谱柱Ultimate C18的色谱图。
图2为实施例2中三氟乙酸体系下色谱柱YMC-Pack Pro C18 RS的色谱图。
图3为实施例2中三氟乙酸体系下色谱柱ACE C18-PFP的色谱图。
图4为实施例2中乙酸铵体系下流动相A的pH为6.7的色谱图。
图5为实施例2中乙酸铵体系下流动相A的pH为9.0的色谱图。
图6为实施例2中乙酸铵体系下流动相A的pH为4.2的色谱图。
图7为实施例2中磷酸体系下色谱柱ACE Excel 2 C18的色谱图。
图8为实施例2中磷酸体系下色谱柱YMC Triart C18的色谱图。
图9为实施例2中辛烷磺酸钠-磷酸二氢钾体系下的色谱图。
图10 为实施例3色谱柱研究的色谱图。
图11为实施例5中梯度1的色谱图。
图12为实施例5中梯度2的色谱图。
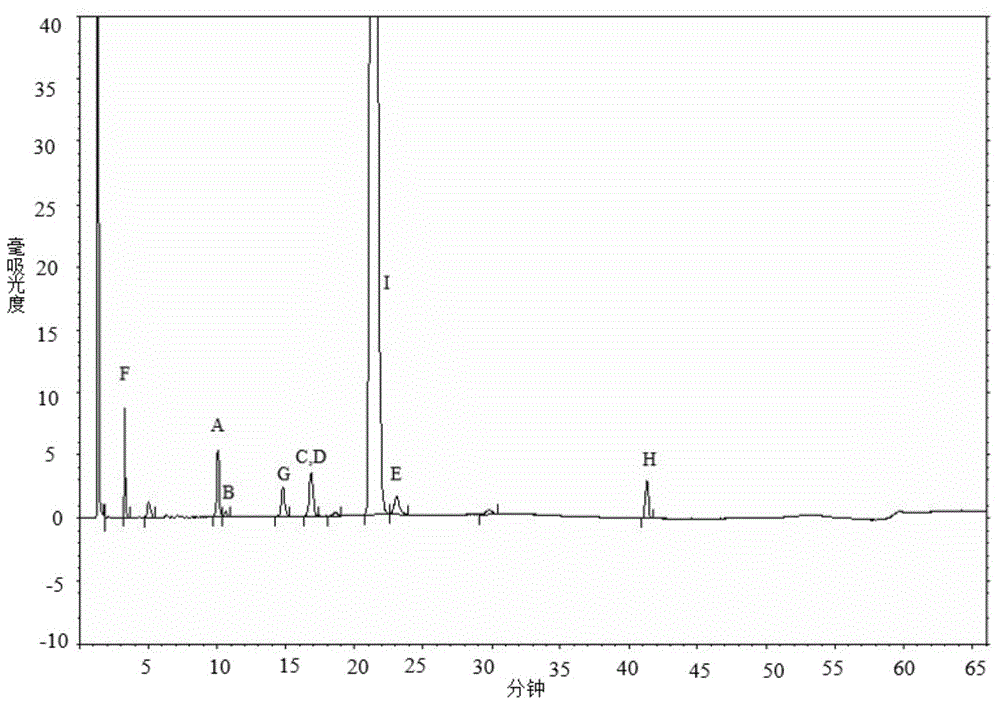
图13为实施例6杂质混合液的色谱图。
具体实施方式
以下结合实施例进一步描述本发明,但这些实施例并非限制本发明的范围。实施例中未注明具体条件的实验方法,通常按照常规条件,或按照原料或商品制造厂商所建议的条件。未注明具体来源的试剂,为市场购买的常规试剂。
实施例1 式(I)马来酸盐的制备
式(I)化合物(按照CN111148515 B的方法制备)中加入乙酸乙酯(0.63 kg)复溶,0.45μm滤膜过滤,用乙酸乙酯(0.4 kg)洗涤,滤液合并至5 L反应釜中。控温20~25 ℃,滴加马来酸(80 g)的乙酸乙酯(2 kg)溶液,滴毕,保温20~25 ℃,搅拌2~3小时。抽滤,用乙酸乙酯(0.8 kg)洗涤滤饼,干燥得白色固体,即式(I)马来酸盐207.28 g。HRMS m/z:405.1779,407.1736 [M+1]+1。H NMR(600MHz, DMSO-d6) δ8.57-8.60(q,1H), 8.50-8.70(brs,1H), 7.74-7.77 (td,J=3.0 Hz,1H), 7.61-7.63(q,J=4.2 Hz,1H), 7.49(m,1H),7.46-7.47(m,1H), 7.44(m,1H), 7.33,7.34(d,J=7.2Hz,1H), 6.03,6.04 (d,J=14.2,Hz,2H), 4.41(s,2H), 3.70-3.73(m,1H),3.63-3.65(m,1H), 3.52-3.60(m,2H), 3.45,3.68(d,J=9.6Hz,2H), 2.77-2.82(m,1H), 2.38-2.40(m,1H), 2.28-2.32(m,1H), 1.97-2.02(m,1H),1.84, 2.39(d,J=13.8Hz,2H), 1.62-1.66(m,1H),1.28-1.32(m,1H),1.06~1.11(m,1H)。
实施例2 流动相体系的研究
2.1试验仪器:高效液相色谱仪:Agilent-1290
2.2溶液的配制
表1 强制降解样品溶液的配制
稀释剂:量取200mL乙腈,800mL水,混合均匀即得。
杂质储备液:精密称取杂质A、B、C、D、E、F、G和H各约19mg,置同一100mL量瓶中,加入适量稀释剂溶解并稀释至刻度,摇匀,即得。其中各杂质浓度为190μg/mL。
分离度溶液:精密称取一定式(I)马来酸盐约32mg置于25mL量瓶中,精密吸取杂质储备液0.25mL至上述量瓶中,加适量稀释剂溶解并稀释至刻度,摇匀,即得。其中式(I)马来酸的浓度约为1.29mg/mL;杂质A、B、C、D、E、F、G和H浓度约为1.9μg/mL。
未破坏供试品溶液:取式(I)马来酸盐约32mg,精密称定,置25mL量瓶中,加10mL稀释剂超声溶解,用稀释剂稀释定容至刻度,摇匀,即得。
2.3试验方法和试验结果
考察不同色谱条件下的分离度,三氟乙酸体系采用强制降解样品溶液和未破坏供试品溶液,乙酸铵体系、磷酸体系、辛烷磺酸钠-磷酸二氢钾体系采用分离度溶液,具体试验条件和试验结果见表2-表5和图1-图9。
2.3.1三氟乙酸体系
表2 三氟乙酸体系色谱条件和试验结果
2.3.2 乙酸铵体系
表3乙酸铵体系色谱条件和试验结果
2.3.3 磷酸体系
表4 磷酸体系色谱条件和试验结果
2.3.4 辛烷磺酸钠-磷酸二氢钾体系
表5 辛烷磺酸钠-磷酸二氢钾体系体系色谱条件和试验结果
试验结果表明,由于杂质多且结构相似,多种流动相体系下主成分与杂质,或各杂质之间的分离度无法满足基本要求,仅辛烷磺酸钠-磷酸二氢钾体系的分离度较高,符合要求。
实施例3 色谱柱的研究
3.1色谱条件
流动相A:0.0025mol/L辛烷磺酸钠(0.01M磷酸二氢钾、0.1%三乙胺,磷酸调pH2.5)溶液;
流动相B:乙腈;
检测波长:210 nm;
流速:1 mL/min;柱温:35℃;进样体积:10μL。
色谱柱:见表6
表6 色谱柱型号
梯度洗脱:见表7
表7 流动相梯度
3.2 试验方法和试验结果
采用实施例2中的分离度溶液,按3.1所述色谱条件,考察三款色谱柱的分离度,试验结果见表8和图10。
表8 三款色谱柱的筛选结果
* RT指保留时间
结果显示:从三款色谱柱的筛选结果可知,杂质A和杂质B、杂质E和主成分化合物(I)较难分离。色谱柱2中杂质E和主成分化合物(I)的分离度小于1.5,色谱柱3可基本实现分离,色谱柱1对两组的分离度均为2.4,较另外色谱柱2和3分离效果好。
实施例4 波长的选择
采用实施例3的色谱条件,色谱柱为Waters Smmetry Shield RP18( 4.6 × 150mm, 3.5μm )。
采用实施例2中的分离度溶液,使用DAD检测器,采用全波长扫描,考察主成分及杂质的最大吸收波长,结果见表9,主成分及杂质的吸收峰均在210nm附近及268nm附近。
表9 主成分、已知杂质波长汇总表
选择主成分和杂质的吸收峰附近210nm和268nm波长考察溶液高温降解及酸降解测定结果,结果对比见表10和表11。
表10 不同波长下溶液高温降解测定结果
RRT:相对保留时间,RT:保留时间
表11 不同波长下酸降解测定结果
结论:杂质在210nm波长下的紫外吸收较268nm强,酸降解样品在210nm的杂质检测个数较268nm的多,优选210nm。
实施例5 洗脱梯度的研究
流动相的梯度洗脱条件如表12所示,色谱柱为Waters Smmetry Shield RP18(4.6 × 150 mm, 3.5μm ),其他色谱条件与实施例3相同,分析强制降解样品溶液(按照实施例2的方法配制)。试验结果见图11和图12。
表12 流动相梯度条件
结论:梯度1的条件下,碱破坏、固体高温及溶液高温条件下的样品30min后仍有降解杂质洗脱出来,分析时间过长。梯度2的条件下,主成分与相邻杂质及各杂质峰之间均能有效分离,且相对于梯度1可以缩短分析时间。
实施例6 专属性试验
6.1 杂质A~H混合液的色谱分析
色谱柱为Waters Smmetry Shield RP18( 4.6 × 150 mm, 3.5μm ),其他试验条件与实施例3相同。
按照实施例2的方法配制分离度溶液,考察分离度。测定结果见表13,色谱图见图13。
表13 杂质A~H混合溶液测定结果
结论:相邻杂质以及各杂质与主成分间的分离度均大于1.5,表明方法对于已知杂质分离度良好。
6.2降解杂质专属性试验
分别在酸、碱、氧化、高温、高湿、光照条件下处理样品供试品,测定其分析结果。
6.2.1 强酸解试验
分别取酸破坏空白、未破坏样品及酸破坏样品进样分析,结果见表14。
表14 酸降解试验样品测定结果
结论:酸降解条件下新增杂质RRT0.21和RRT0.35,已知杂质与相邻杂质、主成分与相邻杂质的分离度均大于1.5,该方法对酸降解样品分离良好。
6.2.2碱解试验
分别取碱降解空白、未破坏样品、碱破坏样品进样分析,结果见表15。
表15 碱降解试验样品测定结果
结论:碱降解条件下新增杂质RRT0.20、RRT0.75、RRT2.12和RRT2.29共4个杂质,已知杂质与相邻杂质、主成分与相邻杂质的分离度均大于1.5,该方法对碱降解样品分离良好。
6.2.3氧化试验
分别取氧化降解空白、未破坏样品、氧化破坏样品进样分析,结果见表16。
表16 氧化试验样品测定结果
结论:氧化降解出12个杂质,已知杂质与相邻杂质、主成分与相邻杂质的分离度均大于1.5,该方法对氧化降解样品分离良好。
6.2.4高温试验
分别取空白、未破坏样品、高温破坏样品进样分析,结果见表17。
表17 高温试验样品测定结果
结论:固体高温下无新增杂质产生,溶液高温下新增杂质RRT2.13。各杂质与相邻杂质、主成分与相邻杂质间的分离度均大于1.5,该方法对高温样品的分离良好。
6.2.5高湿试验
分别取空白、未破坏样品、高湿破坏样品进样分析,结果见表18。
表18 高湿试验样品测定结果
结论:高湿破坏条件下,新增杂质RRT0.25。各相邻杂质、主成分和相邻杂质间的分离度均大于1.5,该方法对高湿降解样品分离良好。
6.2.6光照试验
分别取空白、未破坏样品、光照破坏样品进样分析,结果见表19。
表19 光照试验样品测定结果
结论:已知杂质与相邻杂质、主成分与相邻杂质间的分离度均大于1.5,该方法对各种降解条件的样品分离良好。
- 2,6-二氧杂螺4,5癸烷类衍生物、其制备方法及其在医药上的应用
- 2,6-二氧杂螺4,5癸烷类衍生物、其制备方法及其在医药上的应用