一种基于Tac启动子的质粒表达载体构建及其用途
文献发布时间:2024-04-18 19:53:33
技术领域
本发明涉及一种基于Tac启动子的质粒表达载体改造和构建方法,该方法构建的表达载体在重组蛋白表达中的应用,特别是在人乳头瘤病毒L1抗原蛋白表达及其L1抗原蛋白表达产物在预防宫颈癌中的用途。
背景技术
质粒载体是一种相对分子质量较小、独立于染色体DNA之外的环状DNA(一般大小为1~200kb左右,kb为千碱基对),有的一个细菌中有一个,有的一个细菌中有多个。质粒能通过细菌间的接合由一个细菌向另一个细菌转移,可以独立复制,也可整合到细菌染色体DNA中,随着染色体DNA的复制而复制。
大肠埃希菌(Escherichia coli,俗称大肠杆菌)表达载体最常见的是质粒载体。作为表达载体首先必须满足克隆载体的基本要求,即能将外源基因运载到大肠杆菌细胞中。在克隆载体基本骨架的基础上增加表达元件,就构成了表达载体。各种表达载体的不同之处常常表现于其表达元件的差异。大肠杆菌的质粒表达载体主要包括表达融合蛋白的表达载体和非融合蛋白的表达载体两类。
融合蛋白表达载体是指目的蛋白以融合蛋白的形式表达,并利用载体编码的蛋白或多肽的特殊性质可对目标蛋白进行分离和纯化。与目的蛋白共同表达的融合蛋白部分也被称为标签蛋白或标签多肽(Tag),常用的有谷胱甘肽硫转移酶(glutathione S-transferase,GST)、六聚组氨酸肽(polyHis-6)、蛋白质A(proteinA)和麦芽糖结合蛋白(maltosebinding protein,MBP)等。
融合蛋白表达在载体构建过程中一个关键的问题是两蛋白间的接头序列(Linker),即连接肽,它的长度对蛋白质的折叠和稳定性非常重要。如果接头序列太短,可能影响两蛋白高级结构的折叠,从而相互干扰形不成正确的空间结构;如果接头序列太长,又涉及免疫原性的问题,因为接头序列本身就可能是新的抗原。此外,蛋白药物类或疫苗类产品如果应用融合蛋白表达,由于引入新的外源蛋白或多肽(标签蛋白或切割酶的残留),可能增加药物安全性风险。
表达融合蛋白的载体pGEX是Smith和Johnson于1987年构建的;它的特点是在载体上接上了一种26kDa的谷胱甘肽S-转移酶基因(GST),与其它的融合载体相比,它具有纯化条件温和、步骤简单、无变性剂加入,以及纯化后蛋白能最大限度保持其空间构象和免疫原性的特点;具有较好的应用价值,但载体pGEX编码的GST融合蛋白标签可能会增加药用蛋白产品的安全性隐患。
原核表达载体的重要调控元件包括启动子、SD序列与转录终止子,启动子是DNA链上一段能与RNA聚合酶结合并起始RNA合成的序列,它是基因表达不可缺少的重要调控序列。没有启动子,基因就不能转录。由于细菌RNA聚合酶不能识别真核基因的启动子,因此原核表达载体所用的启动子必须是原核启动子。原核表达系统中通常使用的可调控的启动子有Lac(乳糖启动子)、Trp(色氨酸启动子)、Tac(乳糖和色氨酸的杂合启动子)、lPL(l噬菌体的左向启动子)、T7噬菌体启动子等。
融合蛋白的载体pGEX的启动子是Tac启动子,Tac启动子是一组由Lac和trp启动子人工构建的杂合启动子,受Lac阻遏蛋白的负调节,它的启动能力比Lac和trp都强(表达量高)。其中Tac1是由Trp启动子的-35区加上一个合成的46bp DNA片段(包括Pribnow盒)和Lac操纵基因构成,Tac2是由Trp的启动子-35区和Lac启动子的-10区,加上Lac操纵子中的操纵基因部分和SD序列融合而成。Tac启动子受IPTG的诱导。Tac启动子相对于T7启动子,转录强度较为适中,蛋白表达速度较慢,目的蛋白的折叠速度也较慢,因而其表达的目的蛋白可溶性可能更好。
SD序列(Shine-Dalgarno sequence)是1974年Shine和Dalgarno首先发现,在mRNA上有核糖体的结合位点,它们是起始密码子AUG和一段位于AUG上游3~10bp处的由3~9bp组成的序列。这段序列富含嘌呤核苷酸,刚好与16S rRNA 3’末端的富含嘧啶的序列互补,是核糖体RNA的识别与结合位点。此后人们将此序列命名为Shine-Dalgarno序列,简称SD序列。不同的SD序列及其与起始密码子AUG之间的距离是影响mRNA转录、翻译成蛋白的重要因素之一,某些蛋白质与SD序列结合也会影响mRNA与核糖体的结合,从而影响蛋白质的翻译,也就是影响重组外源基因的表达水平。
用于融合蛋白表达的pGEX载体为美国通用公司(GE)的商业化载体,其启动子为tac启动子,SD序列为AGGAAACAGTA。
通常作为人乳头瘤病毒(Human papillomavirus,HPV)疫苗抗原的HPV-L1蛋白很难用大肠杆菌高效表达HPV-L1可溶性蛋白,其在重组表达时很容易形成包涵体。或者上述蛋白虽然通过包含体纯化,复性等步骤也可得到HPV VLP(Kelsall,S.R.andJ.K.Kulski.Expression of the major capsid protein of human papillomavirustype 16in Escherichia coli.1995.JVirol Methods 53(1):75-90),但是在复性过程中蛋白损失量大,得率低,难以在大规模生产上应用。HPV L1蛋白虽然也可以在大肠杆菌中以正确构象可溶性地表达,溶解于菌体的裂解上清中,但是表达量较低,而且上清中杂蛋白种类多且量大,要从中纯化出目的蛋白难度相当大。通过GST融合表达的方式可以增加上清中L1蛋白的表达量,而且有助目的蛋白的纯化,正如本发明人在专利申请公开文档(CN201410683185)中描述的那样:采用融合蛋白pGEX表达载体可以高效表达人乳头瘤病毒L1蛋白,切除标签蛋白并纯化后可得L1五聚体,能直接用于制备人乳头瘤病毒五聚体疫苗。也可将五聚体高度纯化后,体外组装形成病毒样颗粒(Virus-Like-Particle,VLP),再制备成人乳头瘤病毒VLP疫苗。
但融和蛋白的切割往往需要价格昂贵的酶,同时由于引入新的外源蛋白或多肽(标签蛋白或蛋白酶的残留),可能增加安全性风险。因此,本领域仍然需要成本更低、产量更高、安全性更好的HPV病毒样颗粒(VLP)疫苗生产技术。
发明内容
本发明人经研究出乎意料的发现,pGEX质粒载体固有的SD,不能使HPV16L1或HPV18 L1等外源基因有效地以非融合方式实现可溶表达,即使有表达,其表达量也很低(图2)。在pGEX质粒载体基础上,通过对SD序列的改造、构建了一簇崭新的非融合SD序列表达载体,经转化大肠杆菌后可高效表达可溶性外源蛋白,特别是在人乳头瘤病毒L1抗原蛋白表达方面,其L1蛋白表达量、产量是融合蛋白表达技术的5至10倍(图3),从而使大规模工业化生产宫颈癌疫苗成为可能。本发明基于以上发明现已完成。
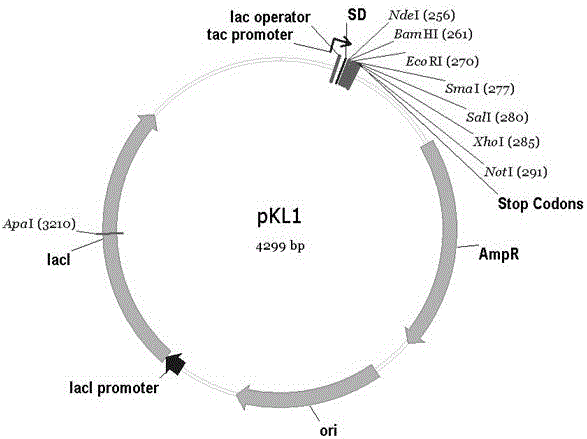
因此,本发明第一方面涉及一簇表达载体的改造,其是基于Tac启动子的质粒表达载体的改造,更具体地是pGEX系列产品的启动子是Tac,pGEX系列产品包括pGEX-1λT、pGEX-2T、pGEX-2TK、pGEX-3X、pGEX-4T-1、pGEX-4T-2、pGEX-4T-3、pGEX-5X-1、pGEX-5X-2、pGEX-5X-3、pGEX-6P-1、pGEX-6P-2、pGEX-6P-3,以及对上述pGEX系列产品进行改造的重组载体。是将pGEX载体中GST融合蛋白编码序列删除,并对SD序列进行转换改造获得。所述表达载体如图1所示。
本发明中所涉及pGEX载体包括pGEX系列产品,例如pGEX-1λT、pGEX-2T、pGEX-2TK、pGEX-3X、pGEX-4T-1、pGEX-4T-2、pGEX-4T-3、pGEX-5X-1、pGEX-5X-2、pGEX-5X-3、pGEX-6P-1、pGEX-6P-2、pGEX-6P-3,以及对上述pGEX系列产品进行改造的重组载体。
本发明中pGEX载体的原SD序列为:AGGAAACAGTA。
在一个实施例中将pGEX-6P-2载体的SD序列转换为AGGAGATATA,同时敲除GST基因,得到的载体称为pKL1;将pGEX-4T-1载体的SD序列转换为AGGAAACAGTA,同时敲除GST基因,得到的载体称为pKL10;将pGEX-2TK载体的SD序列转换为AGGAGGAATAA,同时敲除GST基因,得到的载体称为pKL20;将pGEX-5X-2载体的SD序列转换为AGGAGGAATTA,同时敲除GST基因,得到的载体称为pKL30。
研究中,本发明人还做了载体改造的其他尝试,替换的SD序列列举如下:AGGAAACAGCT,命名为pEZSeq-Kan、替换的SD序列为:AGGAAGCTAAA,命名为pDNR-Dual、替换的SD序列为:AGGAAGACT,命名为pBR322、替换的SD序列为:AGGAGATATACAT,命名为pRSET A、替换的SD序列为:AGGGGTGTT,命名为pSTBlue、替换的SD序列为:AGGAGGTTGT,命名为pEX3。但其效果均不尽人意而舍弃。
本发明第二方面涉及提供一簇重组质粒,包含上述表达载体和插入所述表达载体中的编码外源蛋白的外源基因,外源基因包括但不限于编码微生物例如病毒或者细菌的抗原性蛋白或者多肽的基因、以及编码肿瘤抗原的基因,例如,所述外源基因为人乳头瘤病毒L1蛋白(HPVL1)的DNA序列、人鼻病毒3C蛋白酶(HPV 3C,简称3C酶)编码基因或编码人肠道病毒71型(EV71)衣壳蛋白基因、诺如病毒(NoV)衣壳蛋白基因等等。
本发明第三方面涉及提供一种表达方法,包含宿主细胞及如上所述的本发明的表达载体或者本发明的重组质粒。所述宿主细胞是大肠杆菌,包括但不限于DH5α,GI698,ER2566,BL21(DE3),XA90,B834(DE3)或BLR(DE3)。
本发明第四方面涉及提供一种表达载体的构建方法,为基因重组的方法,包括如下步骤:
S1根据所述SD序列设计突变引物;
S2以pGEX载体为模板进行突变PCR,在原载体中引入NdeI酶切位点;
S3 NdeI/BamHI双酶切切除GST基因序列;
S4 DNA Polymerase I,Klenow片段(克列诺片段,即Large(Klenow)Fragment)补齐粘性末端;所述Klenow片段是DNA聚合酶I C端的一个片段;
S5 T4 DNA ligase连接,获得闭环质粒;
S6设计引物,PCR,在上述质粒中重新引入原酶切位点;
S7序列正确的克隆扩繁,保存菌种,同时提质粒,保存质粒,即获得本发明的载体。
本发明第五方面涉及将本发明的新的SD表达载体应用于多种药用重组蛋白的高效表达,包括可应用于制备人乳头瘤病毒疫苗抗原HPV6、HPV11、HPV16、HPV18、HPV31、HPV33、HPV35、HPV39、HPV45、HPV51、HPV52、HPV56、HPV58、HPV59和HPV68的L1蛋白及病毒样颗粒(VLP),可应用于制备人鼻病毒3C蛋白酶、人肠道病毒71型(EV71)衣壳蛋白及VLP、诺如病毒(NoV)衣壳蛋白及VLP、脊髓灰质炎病毒衣壳蛋白及VLP、甲型肝炎病毒(HAV)衣壳蛋白及VLP和乙型肝炎病毒(HBV)衣壳蛋白及VLP等,在本发明实施前,这些外源蛋白均不能在大肠杆菌细胞中可溶表达,或者可溶表达量很低。
本发明的有益效果:在原核表达系统中,大肠杆菌表达系统具有培养成本低,表达量大的优点。但在大肠杆菌表达系统中表达的HPV L1蛋白往往失去正确天然构象,以包涵体形式表达于沉淀中。目前对表达于包涵体中的蛋白进行复性依然是一个世界性难题。复性困难,效率低下,使得从包涵体中获得有正确构象的VLP在大规模生产中难以实施,只能局限于小规模的实验室研究中。虽然HPV L1也可以正确构象可溶性形式表达于大肠杆菌裂解上清中,但是表达量低下,而且要从大肠杆菌裂解上清中种类繁多的可溶性蛋白纯化出HPV L1蛋白也相当困难,往往需要借助融合表达及亲和层析等手段进行纯化,以上手段往往需要昂贵的酶,及由此导致的药物安全性问题。
本发明采用本发明构建的新型SD质粒表达载体极易于在大肠杆菌表达系统中高效表达HPVL1蛋白,所采用的纯化方法无需使用昂贵的酶,不涉及标签蛋白从而消除了标签蛋白可能引发的药物安全隐患。通过进一步的纯化获得的高纯度HPV L1蛋白可以组装为类病毒颗粒,具有良好的免疫原性,可以诱导高滴度的针对HPV的中和抗体,预防HPV对人体的感染,是一种良好的疫苗形式。
此外,本发明出乎意料的发现,采用上述技术构建的新型SD序列表达载体,经转化大肠杆菌后在人乳头瘤病毒L1抗原蛋白表达方面,其L1蛋白表达量、产量是融合蛋白表达技术的5至10倍,从而使低成本、大规模工业化生产宫颈癌疫苗成为可能。如,用于制备HPV6、HPV11、HPV16、HPV18、HPV31、HPV33、HPV35、HPV39、HPV45、HPV51、HPV52、HPV56、HPV58、HPV59和HPV68的人乳头瘤病毒疫苗L1及VLP抗原蛋白等。
另一方面,本发明构建的新型SD质粒表达载体与适当的宿主细胞配合,可实现多种外源蛋白的高效可溶性表达。如,可应用于制备人鼻病毒的3C蛋白酶表达、人肠道病毒71型(EV71)衣壳蛋白及VLP的表达、诺如病毒(NoV)衣壳蛋白及VLP的表达、脊髓灰质炎病毒衣壳蛋白及VLP的表达、甲型肝炎病毒(HAV)衣壳蛋白及VLP和乙型肝炎病毒(HBV)衣壳蛋白及VLP的表达等。
在参考下列详述和附图后,本发明的这些创造性和新颖性将是显然的。
附图说明
图1表达载体pKL1示意图。而pKL10、pKL20、pKL30质粒图与pKL1类同。
图2.SD序列替换改造前HPV16 L1蛋白表达电泳检测结果,未见L1蛋白表达。显示的是两次实验结果。
图3.SD序列替换改造后HPV16 L1蛋白表达电泳检测结果,L1蛋白大量表达。显示的是两次实验结果。
图4实施例6中HRV 3C蛋白酶大量可溶表达电泳检测结果图。
具体实施方式
本发明提供了一种表达载体,其包含可对多种外源基因进行无融合蛋白表达的全部表达原件。
实施例1、pKL系列载体的构建
1.1通过突变PCR,在pGEX-6P-2质粒中引入NdeI酶切位点:
PCR引物名称及序列如下:
正向引物:6p1-NdeImut-F(5’to3’)
ATTTCA CACAGG AAACAG TACATA TGTCCC CTATAC TAGGTT ATTGGA AAATTA AG;
反向引物:6p1-NdeImut-R序列(5’to3’)
ATAACC TAGTAT AGGGGA CATATG TACTGT TTCCTG TGTGAA ATTGTT ATCC。
PCR反应体系如下:5×phusion HF buffer10μL,ddH
PCR反应程序设置:95℃3min;95℃1min,55℃1min,72℃10min;循环20次;72℃15min。
PCR产物用DpnI消化后转化到大肠杆菌DH5α中,过夜培养后获得单克隆菌落。将单克隆菌落进行扩大培养,之后由专业的基因测序公司对其中的载体序列进行测序,选出测序结果正确的克隆,然后将克隆扩繁并从中提取质粒,获得成功引入NdeI酶切位点的载体。
1.2设计替换SD序列的突变PCR引物,然后通过PCR方法替换原载体的SD序列
引物信息如下:
6PNE-SDm-F(5’to3’):CAATTTCACACAGGAGATATACATATGTCCCCTATACTAGG
6PNE-SDm-R(5’to3’):GTATAGGGGACATATGTATATCTCCTGTGTGAAATTGTTATCC
PCR反应体系如下:5×phusion HF buffer 10μL,ddH
PCR反应程序设置:95℃3min;95℃1min,55℃1min,72℃10min;循环20次;72℃15min。
PCR产物用DpnI消化模板DNA后,转化到E.coliDH5α中,过夜培养后获得单克隆菌落。将单克隆菌落进行扩大培养,之后由专业的基因测序公司对其中的载体序列进行测序,选出测序结果正确的克隆,然后将克隆扩繁并从中提取质粒,获得成功替换SD序列的载体。
1.3载体进行NdeI和BamHI双酶切去除GST基因
酶切体系如下:Cutsmart buffer3μl,ddH
37℃酶切2h;0.8%琼脂糖凝胶电泳,120V,1h;切胶获得去除GST基因后的载体片段对应电泳条带,4℃保存。
用琼脂糖凝胶回收试剂盒回收载体片段,所得载体片段取3μl电泳检测回收结果。然后将双酶切产物用DNA polymerase I补齐粘性末端,反应体系如下:10X T4 DNA Ligasebuffer2.5μl,ddH
将酶切后并末端补齐的载体进行重新连接环化,连接体系如下:T4 DNA Ligasebuffer 2μl,线性平末端载体片段16μl,T4 DNA ligase2μl。16℃连接4h。
连接产物消化后转化到到E.coliDH5α中,过夜培养后获得单克隆菌落。将单克隆菌落进行扩大培养,之后由专业的基因测序公司对其中的载体序列进行测序,选出测序结果正确的克隆,然后将克隆扩繁并从中提取质粒,获得成功替换SD序列并去除GST基因的载体。
1.4PCR扩增质粒,重新引入NdeI和BamHI的酶切位点
PCR引物如下:
6PNE-SDm-noG-F(5’to3’):CAGGAGATATACATATGGGATCCCCGGAATTCCCG
6PNE-SDm-noG-R(5’to3’):GAATTCCGGGGATCCCATATGTATATCTCCTGTGTG
PCR反应体系如下:5×phusion HF buffer10μL,ddH
PCR反应程序设置:95℃3min;95℃1min,55℃1min,72℃10min;循环20次;72℃15min。
PCR产物用DpnI消化模板DNA后,转化到E.coliDH5α中,过夜培养后获得单克隆菌落。将单克隆菌落进行扩大培养,之后由专业的基因测序公司对其中的载体序列进行测序,选出测序结果正确的克隆,然后将克隆扩繁并从中提取质粒,获得成功替换SD序列、去除GST基因,并重新引入NdeI和BamHI的载体。至此,载体pKL1构建完毕。
参照上述实验步骤,以pGEX-2TK为起始载体代替上述实验步骤中的pGEX-6P-2,SEQ IDNo.3为替换的新SD序列,构建pKL20。SD序列突变引物设计如下:
正向引物(5’to 3’)CAATTTCACACAGGAAGGAGGAATAACATATGCCGTCTGAAGCTAC
反向引物(5’to 3’)GACGGCATATGTTATTCCTCCTTCCTGTGTGAAATTGTTATCC
除起始载体及SD序列突变引物外,载体构建其它步骤和方法同pKL1构建。
参照上述实验步骤,以pGEX-5X-2为起始载体,SEQ ID No.4为替换的新SD序列,构建pKL30。SD序列突变引物设计如下:
正向引物(5’to 3’)CAATTTCACACAGGA AGGAGGAATTA CATATGCCGTCTGAAGCTAC
反向引物(5’to 3’)GACGGCATATGTAATTCCTCCT TCCTGTGTGAAATTGTTATCC
除起始载体及SD序列突变引物外,载体构建其它步骤和方法同pKL1构建。
参照上述实验步骤,以pGEX-4T-1为起始载体,SEQ ID No.2为替换的新SD序列,构建pKL10。SD序列突变引物设计如下:
正向引物(5’to 3’)CAATTTCACACAGGA AGGAAACAGTA CATATGCCGTCTGAAGCTAC
反向引物(5’to 3’)GACGGCATATGTACTGTTTCCT TCCTGTGTGAAATTGTTATCC
除起始载体及SD序列突变引物外,载体构建其它步骤和方法同pKL1构建。
至此,含有4种不同SD序列的表达载体(去掉标签的)构建成功,即载体pKL1中的SD序列为AGGAGATATA,pKL10中的SD序列AGGAAACAGTA,pKL20中的SD序列AGGAGGAATAA,pKL30中的SD序列AGGAGGAATTA。
实施例2:基于载体pKL1及pKL30各型HPVL1蛋白的构建
人乳头瘤病毒以下各型外壳蛋白L1基因为人工合成,序列见序列表。具体序列号对应名称如下:
SEQ ID NO:1所示序列为HPV6L1N/C端截短(469aa,Theoretical pI/Mw:5.94/52079.77
Da,N端截短2个氨基酸,C端截短29个氨基酸)
SEQ ID NO:2所示序列为HPV11L1 N/C端截短(470aa,Theoretical pI/Mw:6.08/52274.02Da,N端截短3个氨基酸,C端截短29个氨基酸)
SEQ ID NO:3所示序列为HPV16L1 N/C端截短(472aa,Theoretical pI/Mw:6.08/52548.71Da,N端截短4个氨基酸,C端截短29个氨基酸)
SEQ ID NO:4所示序列为HPV18L1 N/C端截短(473aa,Theoretical pI/Mw:5.94/52722.42Da,N端截短4个氨基酸,C端截短30个氨基酸)
SEQ ID NO:5所示序列为HPV31L1 N/C端截短(473aa,Theoretical pI/Mw:6.27/52869.89Da,N端截短4个氨基酸,C端截短27个氨基酸)
SEQ ID NO:6所示序列为HPV33L1 N/C端截短(471aa,Theoretical pI/Mw:5.88/52765.94Da,N端截短4个氨基酸,C端截短24个氨基酸)
SEQ ID NO:7所示序列为HPV35L1 N/C端截短(470aa,Theoretical pI/Mw:6.10/52494.45Da,N端截短4个氨基酸,C端截短28个氨基酸)
SEQ ID NO:8所示序列为HPV39L1N9 N/C端截短(467aa,Theoretical pI/Mw:6.16/52393.22Da,N端截短9个氨基酸,C端截短29个氨基酸)
SEQ ID NO:9所示序列为HPV45L1 N/C端截短(476aa,Theoretical pI/Mw:5.94/53244.30Da,N端截短4个氨基酸,C端截短30个氨基酸)
SEQ ID NO:10所示序列为HPV51L1t N/C端截短(472aa,Theoretical pI/Mw:5.88/52769.56Da,N端截短4个氨基酸,C端截短28个氨基酸)
SEQ ID NO:11所示序列为HPV52L1 N/C端截短(476aa,Theoretical pI/Mw:5.88/53176.26Da,N端截短4个氨基酸,C端截短23个氨基酸)
SEQ ID NO:12所示序列为HPV56L1t N/C端截短(470aa,Theoretical pI/Mw:6.11/52796.70Da,N端截短4个氨基酸,C端截短25个氨基酸)
SEQ ID NO:13所示序列为HPV58L1 N/C端截短(471aa,Theoretical pI/Mw:5.80/52994.97Da,N端截短4个氨基酸,C端截短23个氨基酸)
SEQ ID NO:14所示序列为HPV59L1 N/C端截短(473aa,Theoretical pI/Mw:6.08/52891.89Da,N端截短4个氨基酸,C端截短31个氨基酸)
SEQ ID NO:15所示序列为HPV68L1a N/C端截短(473aa,Theoretical pI/Mw:5.75/53032.99Da,N端截短4个氨基酸,C端截短28个氨基酸)。
先PCR扩增HPV L1的DNA片段,采用的引物信息如下表1。
表1引物信息
将含有NdeI和Xho1酶切位点的L1基因PCR片段以及重组载体pKL1分别进行NdeI/Xho1双酶切,之后利用T4 DNA连接酶将回收的基因片段与含有对应粘性末端的pKL1进行连接反应,16℃10~15h。连接体系如下:pKL1载体片段6μl,L1基因片段2μl,T4 DNA ligase 1μl,T4 DNA Ligase buffer 1μl。
连接反应后转化连接产物到E.coliDH5α中进行重组子的筛选。将筛选的单克隆菌落进行扩大培养并进行质粒的提取,之后进行测序验证,得到重组表达载体pKL1-HPVL1和pKL30-HPVL1。
将测序结果正确的重组载体化大肠杆菌XA90宿主细胞,并作为表达重组蛋白质的工程菌进行HPV L1蛋白的表达。工程菌培养基为LB培养基(10g/L胰化蛋白胨;5g/L酵母粉;10g/L NaCl)。
0.05%的接种量接种至LB培养基(Amp+)中,于37℃,220rpm培养16h进行活化。取活化后菌液按0.5%的接种量接种至2YT培养基中,于30℃,220rpm培养7h后添加终浓度为0.2mM的IPTG,于30℃,220rpm诱导培养16h后结束发酵,离心收集菌体用于表达量检测以及纯化实验。
SDS-PAGE的结果以HPV16 L1蛋白表达为例,如图2所示,SD序列替换改造前HPV16L1蛋白表达电泳检测结果(未见L1蛋白表达),而图3所示SD序列替换改造后HPV16 L1蛋白表达电泳检测结果(L1蛋白有较大量的表达)。通过量化蛋白表达情况如下表:
表2载体改造前后L1蛋白表达量汇总
从其结果来看,载体改造前基本不表达,采用改造后的载体的蛋白表达量至少达到0.2mg/g湿菌体,最高的可达6.77mg/g湿菌体。
实施例3不同pKL系列载体-HPV16L1表达载体表达HPV16 L1抗原蛋白
将实施例二测序结果正确的重组载体-HPV16L1转化大肠杆菌XA90宿主细胞,并作为表达重组蛋白质的工程菌进行HPV L1蛋白的表达。0.05%的接种量接种至LB培养基(Amp+)中,于37℃,220rpm培养16h进行活化。取活化后菌液按0.5%的接种量接种至2YT培养基中,于30℃,220rpm培养7h后添加终浓度为0.2mM的IPTG,于30℃,220rpm诱导培养16h后结束发酵,离心收集菌体用于表达量检测以及纯化实验。
结果如表3所示,其中可见,采用其中两个SD序列有较大量的表达,而另两个SD序列,则几乎没有表达。
表3含不同SD序列的HPV16L1工程菌目的蛋白表达量对比表
实施例4不同pKL系列载体-HPV59L1表达载体表达HPV59 L1抗原蛋白
将实施例二测序结果正确的重组载体-HPV59L1转化大肠杆菌XA90宿主细胞,并作为表达重组蛋白质的工程菌进行HPV L1蛋白的表达。0.05%的接种量接种至LB培养基(Amp+)中,于37℃,220rpm培养16h进行活化。取活化后菌液按0.5%的接种量接种至2YT培养基中,于30℃,220rpm培养7h后添加终浓度为0.2mM的IPTG,于30℃,220rpm诱导培养16h后结束发酵,离心收集菌体用于表达量检测以及纯化实验。
结果如表4所示,其中可见,采用其中一个SD序列有较大量的表达,而采用另三个SD序列,则几乎没有表达。
表4含不同SD序列的HPV59L1工程菌目的蛋白表达量对比表
实施例5不同pKL系列载体-HPV68L1表达载体表达HPV68L1抗原蛋白
将实施例二测序结果正确的重组载体-HPV68L1转化大肠杆菌XA90宿主细胞,并作为表达重组蛋白质的工程菌进行HPV L1蛋白的表达。0.05%的接种量接种至LB培养基(Amp+)中,于37℃,220rpm培养16h进行活化。取活化后菌液按0.5%的接种量接种至2YT培养基中,于30℃,220rpm培养7h后添加终浓度为0.2mM的IPTG,于30℃,220rpm诱导培养16h后结束发酵,离心收集菌体用于表达量检测以及纯化实验。
结果如表5所示,其中可见,采用其中一个SD序列有较大量的表达,而采用另三个SD序列,则几乎没有表达。
表5含不同SD序列的HPV68L1a工程菌目的蛋白表达量对比表
实施例6:人鼻病毒3C蛋白酶表达载体pKL1-HRV3C的构建
人鼻病毒3C蛋白酶(HRV 3C)基因为人工合成,具体序列如下SEQ ID NO:16。先PCR扩增HRV 3C的DNA片段,引物信息如下:(酶切位点为NdeI和XhoI):
正向引物P3C-NdeI:GGAATTCCATATGGGACCAAACACAGAATTTGCAC
反向引物P3C-XhoI:GGTCTCGAGTTATTGTTTCTCTACAAAATATTG
将含有NdeI和Xho1酶切位点的L1基因PCR片段以及前述改造后的表达载体pKL1分别进行NdeI/Xho1双酶切处理,之后利用T4 DNA连接酶将回收的基因片段与含有对应粘性末端的pKL1进行连接反应,16℃10~15h。
连接体系如下:pKL1载体片段6μl,16L1基因片段2μl,T4 DNA ligase 1μl,T4 DNALigase buffer 1μl。
再次转化鉴定:连接反应后转化连接产物到E.coli DH5α中进行重组子的筛选。将筛选的单克隆菌落进行扩大培养并进行质粒的提取,之后进行测序验证,得到重组表达载体pKL1-HRV3C。
按下述步骤对重组HRV 3C蛋白酶的表达及结果进行检测:
(1)表达载体pKL1-3C转化大肠杆菌宿主菌,同时以空载体pKL1转化宿主菌,作为阴性对照菌株。
(2)表达工程菌单克隆划线及接种:在pKL1-3C转化宿主菌所得的质粒转化平板中,挑选3个克隆,用牙签分别挑取克隆划线传代,然后将划线后的牙签放入40ml含100μg/ml Amp的LB液体培养基中。对照组为从空载体pKL1转化宿主菌所得的转化平板中,选一个取单菌落,划线后,同样接种于40ml含100μg/ml Amp的LB培养基中。培养条件为37℃,220rpm,振荡过夜培养。
(3)乳糖诱导:过夜培养的菌液按0.5%转接2YT发酵培养基,37℃,220rpm,培养至菌液OD600为1.5~2.0时,加入乳糖至终浓度为2g/L,30℃,220rpm条件下进行过夜诱导。
(4)超声波破碎细胞,然后SDS-PAGE电泳检测表达结果,分析目的蛋白是否表达,以及是否为可溶表达。结果见图4(三个单菌落工程菌1#至3#的平行实验),结果显示利用本发明的方法可使HRV 3C蛋白酶大量可溶表达。
- 一种蓝光调节启动子、蓝光调节启动子的融合基因、蓝光介导调节质粒及构建方法和应用
- 一种蓝光调节启动子、蓝光调节启动子的融合基因、蓝光介导调节质粒及构建方法和应用