一种基于基因调控网络构建患者生存网络的方法
文献发布时间:2024-04-18 19:58:21
技术领域
本发明属于分子生物学、系统生物学领域,涉及一种基于基因调控网络构建患者生存网络的方法。
背景技术
在复杂疾病研究中,生存分析被广泛用于鉴定与患者生存和预后相关的疾病标志物,进而指导疾病筛查、早期诊断和个体化医疗决策。传统生存分析主要分为两步:首先根据特定基因的表达水平对患者排序;然后利用对数秩检验评估排名首尾1/2(或1/4)的患者的生存时间是否存在显著差异。与患者生存显著相关的基因被称为癌症生存基因,它们往往与癌症发展和预后密切相关。然而,传统生存分析存在两个局限:
1)利用基因表达水平难以对患者进行准确且稳定的排序。首先,显著的个体差异性导致基因在不同患者体内的表达水平缺乏可比性;此外,复杂的体内和体外因素导致单基因的表达水平缺乏稳定性。
2)基于表达水平难以发现生存相关的调控子(转录因子和小RNA)。首先,很多调控子(特别是miRNA)在肿瘤组织中的表达水平很低,这导致我们难以对它们准确定量并基于它们的表达水平给病人排序;此外,很多调控子通过表达水平变化以外的方式(例如蛋白质结构和微环境)影响靶基因表达,进而影响癌症进展。
基因并非独立发挥功能,而是在复杂的基因调控网络(Gene RegulatoryNetwork,GRN)中相互作用、相互协同。GRN的边代表各种各样的相互作用和功能关联,例如物理相互作用(DNA-DNA相互作用、蛋白质-DNA相互作用、蛋白质-蛋白质相互作用)、遗传相互作用(两个或多个基因关联同一性状)、参与同一生物过程或信号通路等。与基因表达水平相比,GRN具备以下优势:
1)GRN反映了基因在多个患者中稳定的功能关联和调控架构,受个体差异的影响较小;
2)相比单基因表达水平,多基因组成的网络具有更高的数据维度,降低了结果的随机性;
3)基于GRN我们可以忽略调控子的表达水平,而是借助调控子的靶基因逆向推测它与患者生存的关系。
综上,我们相信基于GRN开展生存分析能有效解决传统生存分析的局限,显著拓展癌症预后标志物的发现。
发明内容
针对现有生存分析方法中存在的技术问题,本发明的目的在于提供一种基于基因调控网络构建生存网络的方法。本发明赋予了GRN节点一个新的属性,称为共表达稳定性(co-expression stability)。我们知道,GRN中相互连接的基因往往具有相似的表达模式(表达量在多个样本中同高同低),这种现象称为共表达。共表达的基因往往功能相关或参与同一生物过程。基于这一特点,某个基因在GRN中的共表达稳定性表示该基因与它的所有邻接基因的表达量差异(基于Z-Score标准化保证不同基因表达量的可比性)。表达差异越小,该基因的共表达稳定性越高,此时它与邻接基因组成的功能模块正常运转;表达差异越大,该基因的共表达稳定性越低,此时它与邻接基因组成的功能模块失调。综上,基因的共表达稳定性与它的功能稳定性密切相关,当一个基因在不同患者体内的共表达稳定性与患者的生存时间显著相关时,该基因被认为在癌症进展中扮演重要角色。
基于上述原理,我们建立了基于GRN的生存分析策略。该方法以癌症患者的基因表达数据(微阵列数据、RNA测序数据、蛋白质质谱数据)和生存信息(获取生存信息的手段包括医疗档案和追踪调查等,一些大规模癌症研究项目如TCGA也提供了患者的生存信息)作为输入。主要分析步骤包括GRN构建、共表达稳定性评估、患者排序、以及生存差异评估等。
步骤1)利用实验手段或直接从公共数据库中获取基因表达数据(又称基因表达矩阵,矩阵的行表示所有基因,矩阵的列表示所有患者,矩阵的值表示基因在特定患者中的表达水平,包括转录出的RNA水平或翻译出的蛋白质水平)。实验手段包括基于高通量测序技术检测生物样本中的RNA水平,或基于质谱技术检测生物样本中的蛋白质水平;公共数据库包括Gene Expression Omnibus(GEO)、The Cancer Genome Atlas Program(TCGA)和ArrayExpress等。
步骤2)基于基因表达矩阵构建GRN。现有的GRN推断方法主要包括聚类算法(层次聚类、图聚类等)、机器学习算法(贝叶斯算法、随机森林等)和深度学习算法(卷积神经网络、迁移学习等)。
步骤3)利用实验手段或相互作用数据库优化GRN。目的是删除可信度较低的边,只保留经过实验验证或公共数据库收录的相互作用,从而保证后续分析的准确度。可用于优化GRN的实验手段包括:基于免疫共沉淀预测转录因子-靶基因相互作用,基于酵母双杂交、近距离荧光共振、表面等离子体共振、质谱联用等技术预测蛋白质-蛋白质相互作用。可用于优化GRN的相互作用数据库包括:染色质相互作用数据库(4DGenome),转录因子-靶基因数据库(TRRUST和hTFtarget),小RNA-靶基因数据库(miRDB和miRTarBase),蛋白质-蛋白质相互作用数据库(STRING和HuRI)和通路数据库(KEGG和Reactome)。
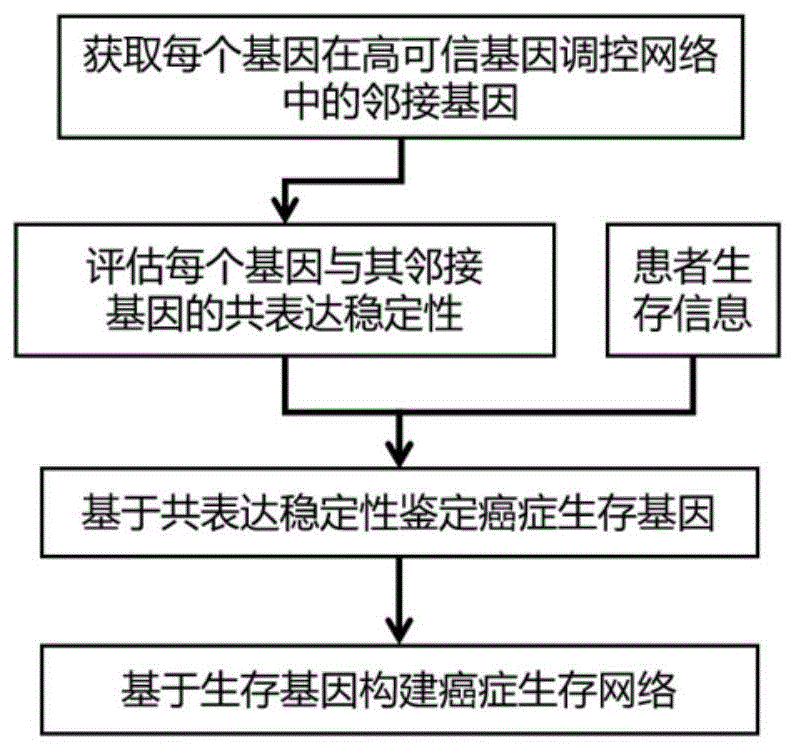
步骤4)评估GRN中每个节点(每一节点对应一基因)在不同患者中的共表达稳定性。具体步骤包括先获取每个基因在GRN中的邻接基因;对每个基因在所有患者的表达水平进行Z-Score标准化,目的是保证不同基因的表达水平具有可比性;基于每个基因的邻接基因评估其共表达稳定性(详见“具体实施方式”)。
步骤5)针对所述基因表达矩阵中的每一基因,基于该基因在各患者中的共表达稳定性对患者排序,取共表达稳定性排名前1/4和后1/4的两组患者的生存信息进行Kaplan-Meier生存分析并得到该基因的对数秩检验P值,然后基于该P值评估两组患者的生存时间是否具有统计学差异。P≤0.05时具有统计学差异,表明该基因的共表达稳定性显著影响患者生存时间;P>0.05则表明该基因的共表达稳定性不影响患者生存时间。
步骤6)保留对数秩检验P≤0.05的基因以及GRN中连接这些基因的边及该边连接的基因,利用cytoscape工具构建目标癌症的生存网络。该网络中,相连节点的共表达水平在不同患者间发生扰动,而这些扰动会显著影响患者的生存时间,具有重要的研究价值。
基于上述内容,本发明的技术方案为:
一种基于基因调控网络构建患者生存网络的方法,其步骤包括:
1)获取基因表达矩阵,所述基因表达矩阵的行为基因,所述基因表达矩阵的列为目标癌症患者样本,所述基因表达矩阵中第m行第n列的元素值表示第m个基因在第n个目标癌症患者中的表达水平;获取每一所述目标癌症患者样本对应的患者生存信息;
2)基于所述基因表达矩阵构建基因调控网络;
3)对于所述基因调控网络中的每一条边,如果该边的可信度低于设定阈值,则删除该边;
4)评估步骤3)优化后的基因调控网络中每个基因在每一目标癌症患者样本中的共表达稳定性;
5)对于所述基因表达矩阵中的每一基因,基于该基因在各目标癌症患者样本中的共表达稳定性对各目标癌症患者样本排序,取共表达稳定性排名前T%的目标癌症患者样本的生存信息作为第一组信息,取共表达稳定性排名后T%的目标癌症患者样本的生存信息作为第二组信息;基于第一、二组信息进行生存分析得到该基因的对数秩检验值P;然后基于该基因的对数秩检验值P判定该基因对该排名前T%的目标癌症患者样本、后T%的目标癌症患者样本中各目标癌症患者的生存时间是否具有统计学差异;如果具有统计学差异,则保留该基因;
6)根据步骤5)中所保留的基因及所述基因调控网络中连接各所保留基因的边和基因,
构建目标癌症的生存网络。
进一步的,得到每个基因在各目标癌症患者样本中的共表达稳定性的方法为:首先获取基因调控网络中每个基因的邻接基因;然后获取每一所述邻接基因在所述基因表达矩阵中所有患者的表达水平并对其进行Z-Score标准化;然后基于每个基因的Z-Score标准化的邻接基因评估该基因在各目标癌症患者样本中的共表达稳定性。
进一步的,得到每个基因在各目标癌症患者样本中的共表达稳定性的方法为:对于所述基因表达矩阵中的每一基因g
进一步的,如果基因的对数秩检验值P≤0.05,则判定该基因对该排名前T%的目标癌症患者样本、后T%的目标癌症患者样本中各目标癌症患者的生存时间具有统计学差异。
进一步的,利用实验手段或相互作用数据库优化所述基因调控网络,删除所述基因调控网络中可信度低于设定阈值的边。
进一步的,所述实验手段包括:基于免疫共沉淀预测转录因子-靶基因相互作用、基于酵母双杂交、近距离荧光共振、表面等离子体共振、质谱联用;所述相互作用数据库包括:染色质相互作用数据库、转录因子-靶基因数据库、小RNA-靶基因数据库、蛋白质-蛋白质相互作用数据库和通路数据库。
一种服务器,其特征在于,包括存储器和处理器,所述存储器存储计算机程序,所述计算机程序被配置为由所述处理器执行,所述计算机程序包括用于执行上述方法中各步骤的指令。
一种计算机可读存储介质,其上存储有计算机程序,其特征在于,所述计算机程序被处理器执行时实现上述方法的步骤。
本发明具有以下优势:
1)解决了传统生存分析稳定性不足的问题。相比基因表达水平,基因在GRN中的拓扑特征能更稳定地反应患者的生理状态。首先,GRN反映了基因在多个患者中稳定的功能关联和调控架构,因此受个体差异的影响较小;此外,相比单基因表达水平,多基因组成的网络具有更高的数据维度,降低了结果的随机性。
2)能基于生存基因逆向推导有驱动癌症进展的调控子(转录因子和小RNA)。我们知道,基于新方法获取的生存基因在不同患者体内的共表达水平不同。而调控子是造成靶基因共表达的主要原因之一。换言之,调控子是否发挥作用会造成靶基因在不同患者中共表达水平不同,进而影响患者生存。因此,生存基因靶向的转录因子或小RNA与癌症进展和患者生存密切相关。调控的生存基因越多,转录因子或小RNA在癌症中的角色越重要,可信度越高。
附图说明
图1为本发明的构建基因调控网络流程图。
图2为本发明的构建患者生存网络流程图。
图3为本发明的算法示意图。
具体实施方式
下面将结合附图和具体实施方式对本发明做进一步的说明。
本发明的流程如图1、图2所示,假设某一目标癌症的基因表达数据包含m个不同的基因和n个病人样本,另外获取了这n个病人的生存信息,下面将结合附图和具体实施方式对本发明做进一步的说明。
步骤1)基于生物信息学手段构建这m个基因的基因调控网络。可用的GRN推断方法包括聚类算法、机器学习算法和深度学习算法。聚类算法包括层次聚类(WGCNA)、图聚类(MCL)等;机器学习算法包括贝叶斯算法(BANJO、CLR)、随机森林(GENIE3、ReNI)等;深度学习算法包括卷积神经网络(DeepInsight、DeepFeature)、迁移学习(Geneformer)等。
步骤2)利用实验手段或相互作用数据库优化GRN,保留可信度较高的相互作用。实验手段包括基于免疫共沉淀预测转录因子-靶基因相互作用,基于酵母双杂交、近距离荧光共振、表面等离子体共振、质谱联用等技术预测蛋白质-蛋白质相互作用。公共数据库包括染色质相互作用数据库4DGenome,转录因子-靶基因数据库TRRUST和hTFtarget,小RNA-靶基因数据库miRDB和miRTarBase,蛋白质-蛋白质相互作用数据库STRING和HuRI,通路数据库KEGG和Reactome。
步骤3)获取GRN中每个基因的邻接基因。基因与其邻接基因可以通过各种相互作用连接,包括物理相互作用(DNA-DNA相互作用、蛋白质-DNA相互作用、蛋白质-蛋白质相互作用)、遗传相互作用(两个或多个基因关联同一性状)、以及共调控(靶向同一转录因子或小RNA,或参与同一生物过程或信号通路)。
步骤4)对基因在不同患者中的表达水平执行Z-Score标准化。目的是消除不同基因表达量之间的差异,只保留基因在不同患者中的相对变化,从而使不同基因的表达水平具有可比性。
步骤5)评估每个基因与其邻接基因的共表达稳定性。共表达稳定性表示一个基因与其所有邻接基因的相关程度,它侧面反应了该基因的功能稳定性。假设基因g
步骤6)基于共表达稳定性的生存分析。对于基因表达矩阵中的每个基因,基于其在每个患者中的共表达稳定性对患者排序,对共表达稳定性排名前1/4和后1/4的两组患者进行Kaplan-Meier生存分析并得到对数秩检验P值,然后基于该P值评估两组患者的生存时间是否具有统计学差异。P≤0.05时具有统计学差异,表明该基因的共表达稳定性显著影响患者生存时间;P>0.05则表明该基因的共表达稳定性不影响患者生存时间。
如图3所示,上方路径表示传统生存分析算法,基于基因g
步骤7)构建癌症生存网络。保留对数秩检验P≤0.05的基因以及GRN中连接这些基因的边,组成目标癌症生存网络。该网络中,相连节点的共表达水平在不同患者间发生扰动,而这些扰动会显著影响患者的生存时间,具有重要的研究价值。
综上,针对传统生存分析的不足,本发明赋予了GRN节点一个新的属性——共表达稳定性,并建立了共表达稳定性与患者生存的关联。值得一提的是,新方法与传统方法发现的生存基因作用机制不同:传统生存基因通过自身表达水平影响患者生存,而我们的生存基因通过在GRN中的扰动影响患者生存。因此,新方法的意义不在于取代传统生存分析方法,而是从新的维度拓展癌症生存基因的发现,与传统方法形成良性互补。
随着癌症精准医学研究的深入,癌症标志物的发现进入平台期。亟需从不同维度研究基因与癌症进展和患者生存的关系。因此,基于GRN的生存分析策略具有广阔的应用前景。
尽管为说明目的公开了本发明的具体实施例,其目的在于帮助理解本发明的内容并据以实施,本领域的技术人员可以理解:在不脱离本发明及所附的权利要求的精神和范围内,各种替换、变化和修改都是可能的。因此,本发明不应局限于最佳实施例所公开的内容,本发明要求保护的范围以权利要求书界定的范围为准。
- 一种基于集成特征重要性和鸡群算法的基因调控网络构建方法
- 一种基于动态贝叶斯网络的基因调控网络构建方法
- 一种基于元分析构建基因调控网络的方法