制备L-草铵膦的方法
文献发布时间:2024-04-18 19:58:21
发明领域
本发明涉及一种制备并得到除草剂草铵膦(glufosinate)的L-异构体的方法。本发明适合由D,L-草铵膦的外消旋混合物制备并得到该L-异构体。本发明尤其适合优化由D,L-草铵膦的外消旋混合物得到L-异构体的产率。
此外,本发明通常还适用于旨在将酸性化合物的烷基铵盐转化为相应铵盐的方法。
背景
具有IUPAC名称:(2RS)-2-氨基-4-[羟基(甲基)膦酰基]丁酸或4-[羟基(甲基)膦酰基]-DL-高丙氨酸(CAS登记号51276-47-2)和通用名DL-4-[羟基(甲基)膦酰基]-DL-高丙氨酸盐的除草剂草铵膦是叶面施用的非选择性除草剂并且从毒理性或环境角度被认为是最安全的除草剂之一。
草铵膦铵盐(IUPAC名称:(2RS)-2-氨基-4-(甲基次膦酸酯基)丁酸铵,CAS登记号77182-82-2)尤其是众所周知的其农学上可接受的盐。
草铵膦铵盐及其除草活性例如也已由F.Schwerdtle等人,Z.Pflanzenkr.Pflanzenschutz,1981,Sonderheft IX,第431-440页描述。
Hoerlein等人在Rev.Environ.Contam.Toxicol.(第138卷,1994)“草铵膦(膦丝菌素),一种具有出人意料的除草性能的天然氨基酸”中讨论了该谷氨酸合成抑制剂草铵膦。
US 4,168,963描述了具有除草活性的含磷化合物,其中膦丝菌素(2-氨基-4-[羟基(甲基)膦酰基]丁酸,通用名:草铵膦)及其盐尤其已经在农化(农业化学)领域中获得商业重要性。
草铵膦是外消旋体并且由下列结构(1)表示:
其他草铵膦是两种对映体的外消旋体,这两种对映体中仅一种显示出足够的除草活性(例如见US 4265654和JP92448/83)。L-草铵膦比D-草铵膦有效得多(Ruhland等人(2002)Environ.Biosafety Res.1:29-37)。
目前用于草铵膦的商业化学合成方法得到L-和D-草铵膦的外消旋混合物(Duke等人,2010Toxins 2:1943-1962)。
呈外消旋体的草铵膦及其盐以牌号Basta
具有IUPAC名(2S)-2-氨基-4-[羟基(甲基)膦酰基]丁酸(CAS登记号35597-44-5)并且也称为草铵膦-P的L-草铵膦可以市购或者例如可以如US 10260078B2,WO 2006/104120,US 5530142,EP 0248357A2,EP 0249188A2,EP 0344683A2,EP 0367145A2,EP0477902A2,EP 0127429和J.Chem.Soc.Perkin Trans.1,1992,1525-1529所述制备。
然而,基于常规,即动态动力学拆分外消旋体(如在WO18108794中)或酶催化外消旋体拆分(如在EP0054897中例如用PG酰胺酶)或者基于不对称合成如不对称氢化(如在EP0238954或EP1864989中)的对映体选择性合成要求额外的助剂如难以得到的原料或其他助剂或者要求额外的合成步骤,这使得该合成更为复杂并且因此更为昂贵。因此,这些方法均尚未证明与合成外消旋材料相比具有成本竞争力。
发明概述
因此,鉴于需要活性对映体L-草铵膦,仍然需要主要,甚至优选仅生产活性L-形式的方法。
此外,还需要以简单且成本有效的方式尤其得到草铵膦铵盐,其为一种成熟、已经注册并且农业上可靠的盐。
本文描述了生产L-草铵膦,尤其是得到其铵盐的新且成本有效的方法。
详细描述
草铵膦最初以外消旋混合物合成。市场上大多数包含草铵膦的除草产品包含L-草铵膦和D-草铵膦的50:50混合物,然而观察到的除草活性如上所述由L-草铵膦完成,而D-草铵膦没有活性并且反而将没有除草活性的手性化合物释放到环境中。
因此,需要仅提供,或者至少主要提供草铵膦的活性L-异构体的有效、简单且成本有效方法。
现有技术中得到L-草铵膦的一种方法是通过D,L-草铵膦的去外消旋化。
例如,WO 2017151573描述了式DL-(1)的外消旋草铵膦铵盐在两步方法中的去外消旋化(方案1),其中第一步涉及使D-草铵膦氧化脱氨成PPO(2-氧代-4-(羟基(甲基)膦酰基)丁酸)并且第二步涉及使用来自一种或多种胺供体的胺基团将PPO特异性胺化为式L-(1)的L-草铵膦铵盐。
方案1:
然而,尽管据说通过将这两个反应组合可以由外消旋草铵膦混合物开始显著增加L-草铵膦的比例,即得到基本由L-草铵膦构成的组合物,但该方法仍有一个缺点需要注意。
该反应的全貌表明式L-(1)的L-草铵膦铵盐不以化学计量比得到,而是作为副产物也得到L-草铵膦的胺供体铵盐。
例如,若该胺供体如方案1a所示为异丙胺,则这得到盐的混合物,即式L-(1)的所需L-草铵膦铵盐和未登记的式L-(2)的L-草铵膦异丙铵盐的混合物。
方案1a:
因此,尽管所得L-草铵膦铵盐L-(1)随后可以提纯或基本提纯并且用作除草剂,但式L-(2)的L-草铵膦异丙铵盐需要进一步处理以也从其受益并且也得到所需L-草铵膦铵盐。
L-草铵膦副产物的进一步处理通常取决于其性质有可能在使该方法毫无吸引力的程度上变得繁琐、复杂和耗时。
正如WO 2017151573所述,合适胺供体的选择对于将D-草铵膦经济地转化为L-草铵膦而言是重要的。列出了各种问题,如该供体的可得性和成本,该供体的潜在回收以及副产物,例如酮基副产物与所需L-草铵膦的分离。
WO 2017151573推荐可以使用接受几种不同胺供体的转氨酶,包括低成本胺供体如苯基乙胺、L-天冬氨酸或外消旋天冬氨酸、L-谷氨酸或外消旋谷氨酸、L-丙氨酸或外消旋丙氨酸、仲丁胺和异丙胺。后者据说任选是有利的,因为从L-草铵膦除去副产物丙酮可以驱使该反应完全。然而,仍要面对几个问题,有时是后续的问题:
例如,若胺化的副产物,例如由该供体分子得到的酮基化合物不能从反应混合物连续除去,则首先必须大大过量使用胺供体以改变反应平衡而驱使所需反应完全,其次随后从所需L-草铵膦除去过量胺供体要求额外的提纯步骤。
然而,即使由供体分子得到的酮基化合物可以容易地从反应混合物连续除去,就像由异丙胺得到丙酮的情况一样,还将面临其他后续问题。
例如,若将烷基胺用作该供体,则该烷基胺代表比氨更强的碱并且因此作为不希望的副反应观察到由起始铵盐置换氨。这导致形成L-草铵膦的烷基铵盐,后者由于未获得监管机构的批准而不能用于农业目的和商业组合物。
有鉴于此,在现有技术中选择的优选氨基供体是谷氨酸,因为由氨基转移反应得到的酮酸副产物,即α-酮基戊二酸,可以用成熟和已知的方法分离和/或提纯并且可以进一步用于许多应用中,包括用于合成药物制剂、食品添加剂和生物材料。进一步提到可以将其化学转化为外消旋谷氨酸或L-谷氨酸,它们任选再用于该反应中,并且化学还原胺化涉及将酮基转化为胺。
然而,尽管已提及,但并不认为异丙胺是优选的胺供体。
此外,如上所示,找到一种可以立即使用的野生型转氨酶显然并不容易。非常经常的是该类野生型转氨酶通常不接受所需胺供体并且将需要进行进化以最终接受所需底物,这可能提出进一步的挑战。
鉴于上述问题,待解决的问题是两个:首先从该混合物有效分离L-草铵膦的不想要的胺供体盐,然后额外地—甚至更好地—将其直接转化为L-草铵膦的铵盐L-(1)并由此增加所需铵盐的产率。
本发明的解决方案涉及一方面正确选择胺供体以及除去该胺供体的经济且环境友好步骤,由此得到更高化学计量产率的所需L-草铵膦铵盐。
现在惊人地发现一种新方法,其允许以容易、简单、环境和成本有效的方式由D,L-草铵膦外消旋体得到L-草铵膦,以及尤其是L-草铵膦的有利地期望的铵盐。
因此,发现首先需要从脂族C
选择该类脂族C
方案1.A:
因此,式L-(2)中的R
优选该脂族C
在氨基转移之后接下来的挑战是要成功地将不想要的副产物L-草铵膦C
该问题可以通过在第一中间步骤中加入Ca(OH)
方案2-1:
由于是较弱的碱,该L-草铵膦C
在第二中间步骤中,将在水溶液中离解的硫酸铵加入式L-(3)的L-草铵膦钙盐的剩余水溶液中,硫酸根离子形成沉淀的水不溶性硫酸钙(CaSO
方案2-2
石膏是无害、无毒和自身安全的材料,可以通过过滤简单除去。可以通过除去溶剂而由现在含有式L-(1)的所需L-草铵膦铵盐的剩余水溶液得到纯盐并且铵盐L-(1)以几乎定量产率得到。
本发明的实施方案
在下文给出将D-草铵膦转化为L-草铵膦的方法的单个和优选实施方案。
这些实施方案包括将D-和L-草铵膦的外消旋混合物的低成本原料转化为除草产品的有利和优选方式,其中已经显著富含除草活性的L-对映体L-草铵膦。
本发明方法包括转化的方式和方法以及富集所需L-对映体的份额及其分离的方式。
这些方法包括几个步骤,这些步骤可以在单一容器中连续进行(一锅法)或者在几个分开的容器中连续进行(多锅法)。
DAAO酶
第一步是使D-草铵膦(可以存在于D-和L-草铵膦的外消旋混合物中)氧化脱氨成PPO(2-氧代-4-(羟基(甲基)膦酰基)丁酸)。
方案1-1
该步骤优选由D-氨基酸氧化酶(DAAO)催化。
然而,使D-草铵膦氧化脱氨成PPO可以由几种酶催化,但也可以非酶促发生。可能的酶包括DAAO、DAAD(D-氨基酸脱氢酶(DAAD)酶)和D-氨基酸脱水酶。
在一个实施方案中,使用DAAO酶催化D-草铵膦向PPO转化。该反应具有下列化学计量:
D-草铵膦+O
由于氧气在含水反应缓冲液中的溶解度与草铵膦相比通常要低,对于有效方法而言必须在该DAAO反应的整个时间段内引入氧气。
D-草铵膦最初以大于30g/L至高达140g/L存在。
氧气通常最初以大约8mg/L存在,但在整个反应过程中加入以允许有足够的氧气使反应继续快速进行。水通常,但非强制性地,以大于500g/L存在。
几种DAAO酶在本领域中是已知的并且可以用于本文所述的方法中,只要它们能够接受D-草铵膦作为底物并且提供足以达到驱动该反应的水平的活性。可以用于该方法中的该类DAAO酶包括来自圆红冬孢酵母(Rhodosporidium toruloides),变异三角酵母(Trigonopsis variabilis),镰孢菌属(Fusarium sp),假丝酵母属(Candida sp),裂殖西葫芦菌属(Schizosasaccharomyces sp),轮枝孢属(Verticillium sp),洁丽新韧伞(Neolentinus lepideus),里氏木霉(Trichoderma reesei),油脂丝孢酵母(Trichosporonoleaginosus)等的已经修饰以提高活性的那些。
特定起始酶已经在WO2017/151573中描述并鉴定,其作为参考整体引入本文,在这里尤其是由第7页开始。
额外的DAAO酶可以以各种方式鉴定,包括序列相似性和功能筛选。这里若DAAO酶是突变DAAO酶,则它需要能够接受D-草铵膦作为底物。其他DAAO酶可以类似地修饰以接受D-草铵膦并且具有更大的活性。以相同方式可以通过诱变改进已知DAAO酶和/或可以鉴定新的DAAO酶。
在一些实施方案中,可以在本文所述的方法中制备和测试突变酶。突变DAAO酶(例如来自薄红酵母(Rhodotorula gracilis))可以与野生型序列相比在突变序列中的位置处包括一个突变、两个突变、三个突变或者多于三个突变(例如四个突变、五个突变、六个突变、七个突变、八个突变、九个突变或十个突变或者更多个)。这里还参考WO2017/151573中的公开内容。
其他合适的D氨基酸氧化酶可以由真菌来源得到。
如所示那样,可以鉴定和测试DAAO酶以用于本发明方法中。为了确定该酶是否接受D-草铵膦作为底物,可以使用产物形成的氧电极测定(Hawkes,2011)、比色测定(Berneman A,Alves-Ferreira M,Coatnoan N,Chamond N,Minoprio P(2010),测定D-氨基酸的中/高通量D-氨基酸氧化酶比色法.氨基酸外消旋酶的应用.J Microbial BiochemTechnol 2:139-146)和/或直接测量(经由高效液相色谱法(HPLC)、液相色谱质谱法(LC-MS)或类似方法)。
由该DAAO酶催化的反应需要氧气。在一些实施方案中,在液面上空间或者通过将气体喷射通过反应容器将氧气、富氧空气、富氧气流或空气引入该反应中,以提高反应速率。
当DAAO酶催化D-草铵膦向PPO转化时,形成过氧化氢(H
在一些实施方案中,可以使用催化和非催化分解反应消除过氧化氢。例如,过氧化氢可以通过使用增加的热量和/或pH的非催化分解反应消除。过氧化氢还可以通过使用例如过渡金属和其他试剂,如碘化钾的催化分解反应消除。除了消除过氧化氢外,过氧化氢酶的使用还产生氧气(O
可以使用其他酶来催化D-草铵膦向PPO转化。例如,可以以下列化学计量使用接受D-草铵膦作为底物的DAAD酶:D-草铵膦+H
认识到在其中使用DAAD的方法中,该DAAD催化的反应可以包括氧化还原辅因子再循环。这涉及氧化该已还原受体,因而它可以接受来自D-草铵膦的更多电子。
D-草铵膦基本完全(大于80%,大于85%,大于90%或大于95%或者80-100%,85-99%,90-99%或95-99%)转化为PPO可以在24小时、18小时、12小时或8小时内发生。
TA酶
第二步为使用来自一种或多种胺供体的胺基团由转氨酶(TA)将PPO特异性胺化为式L-(1)的L-草铵膦铵盐和式L-(2)的烷基铵盐。
方案1-2
该第二步骤涉及使用例如转氨酶(TA)酶、L-氨基酸脱氢酶(LAAD)酶或者通过化学转化将PPO转化为L-草铵膦。
在一个优选实施方案中,该方法为由TA催化的反应。
转氨酶(TA)通常是生产用于药物和精细化学工业的手性胺的重要酶。已经使用一系列方法发现用于这些工业中的新型TA酶,这些方法包括活性引导的方法以及从培养微生物的同源序列搜索到使用关键基序的搜索和环境DNA文库的宏基因组挖掘(Kelly等人(2020)Appl Microbiol Biotech104:4781-4794)。
若该反应作为其中在不存在胺供体和/或转氨酶下将D-草铵膦基本转化为PPO的两步法进行,则PPO在第二步中的起始量通常在30-140g/L范围内。若该反应以单步法进行,则PPO的起始量通常小于1g/L并且在该反应过程中PPO的最高水平通常小于25g/L。该胺供体相对于起始量的外消旋草铵膦最初以1-6摩尔过量存在。
可以用于本文所述方法中的TA包括来自大肠杆菌(Escherichia coli)(UniProtP22256)的gabT转氨酶,已经显示其催化与PPO作为底物的所需反应(Bartsch等人(1990)Appl Environ Microbiol.56(1):7-12)。已经进化出另一种酶以使用异丙胺作为胺供体以较高速率催化所需反应(Bhatia等人(2004)Peptide Revolution:Genomics,Proteomics&Therapeutics,Proceedings of the Eighteenth American Peptide Symposium,编辑Michael Chorev和Tomi K.Sawyer,2003年7月19-23日,第47-48页)。额外地,来自许多微生物,如吸水链霉菌(Streptomyces hygroscopicus)、产绿色链霉菌(Streptomycesviridochromogenes)、白假丝酵母菌(Candida albicans)等的TA酶可以用于实施本文所述方法。
需要的话,这些酶也可以通过诱变进行进化以增加其活性。可以通过Schulz等人,Appl Environ Microbiol.(1990)Jan.56(1):1-6所述的测定法和/或通过由HPLC、LC-MS直接测量产物或类似产物对所需活性选择突变TA酶。
用于这些方法中的额外TA酶可以通过对所需活性筛选TA的集合,如ProzomixLimited(英国Northumberland),SyncoZymes(中国上海),Evocatal(德国Monheim amRhein),Codexis(加拿大Redwood City)或Abcam(英国Cambridge)销售的那些而鉴定。替换地,序列相似性可以用来鉴定新型TA酶。
最后,TA酶还可以从能够催化所需反应的生物鉴定并且甚至可以通过酶工程进一步修饰和优化以允许具有更好选择性和更高输出量的经济生产。
后者例如在作为参考整体引入本文的WO 2020/025577中完成,其描述了具有改进ω-转氨酶(ω-TA)活性的蛋白质,编码具有改进ω-TA活性的相应蛋白质的核酸分子以及立体选择性合成手性胺和氨基酸或增加对映体混合物中的手性胺异构体的方法。尤其提供了与相应野生型ωTA相比具有改进的反应动力学、改进的底物接受度和改进的比活性并且因此使得能够开发胺化产物的经济有效生产方法的在其氨基酸序列中包含修饰的ω-转氨酶(ω-TA)变体。这些变体可以产生磷酸氨基酸如草铵膦的对映体富集、近乎纯净或纯净化合物。
WO2020025577中优选的ω-TA变体允许生产以对映体过量包含(S)-胺的组合物,因此尤其适合以对映体过量得到(S)-草铵膦,其中用于草铵膦的术语(S)-对映体确定了相同的所需L-草铵膦。方便的是相应ω-TA变体同时降低该组合物中(R)-对映体的量,因此得到的草铵膦的无活性D-对映体更少。
PPO基本完全转化为L-草铵膦可以在24小时、18小时、12小时或8小时内发生。就此而言,基本完全是指PPO向L-草铵膦的转化率大于约80%,大于约85%,大于约90%,大于约95%,大于约98%或大于约99%(或者约80-100%,约85-100%,约90-100%或约95-99%)。
胺供体
胺供体分子是包含胺基团的分子,其将胺基团贡献给胺受体分子,因此胺供体的胺基团变为羰基。
在本发明中选择的胺供体分子是C
如上面进一步所述,胺供体要选自脂族C
优选胺供体要选自脂族仲C
因此,在本发明的一个实施方案中,该脂族仲C
在本发明的更优选实施方案中,该脂族仲C
尤其优选该脂族仲C
方案1a:
Kelly等人(2017,Chem.Rev.2018,118,1,349-367)例如描述了已经证明丙氨酸作为TA催化反应的胺供体就其被酶广泛接受以及除去丙酮酸盐副产物的各种方案而言很受欢迎。然而,指出了使用丙氨酸导致不利的反应平衡,其远在原料这侧。后来Kelly等人((2020)Appl Microbiol Biotech 104:4781-4794)描述了具有遍及已知S-选择性TA的那些中良好分布的序列的TA的进化多样性。这些中的一种(称为“pQR2189”)显示出接受氨基供体异丙胺(“IPAm”)的能力。
异丙胺的优点不仅在于其可接受的化学品价格和可以容易地除去副产物,而且在于改进转化率的显著进步。
当在本发明方法中用作氨基供体分子时,异丙胺通过ω-TA的作用转化为丙酮。丙酮是挥发性化合物,产生的优点是它在较低温度下蒸发。这允许在发生反应的过程中从反应混合物除去该ω-TA产生的丙酮,产生的有利效果是该反应的平衡转向由本发明生产胺的方法生产的所需胺。这允许大量得到所需胺,因为由ω-TA催化的逆反应由于缺乏一个反应参与物而降低。
前面提到的WO2020025577也涉及用作氨基供体分子的烷基胺如2-丁基胺和异丙胺。
异丙胺作为TA的胺供体的作用也可以在其他出版物如Park等人(2013,Organic&Biomolecular Chemistry 11,6929-6933)中找到,其公开了不同转氨酶在通过使用异丙胺和各种其他化合物作为胺供体由酮酸对映体选择合成非天然氨基酸中的性能。
优选用于产生胺的胺供体分子以10-250g/L(克/升),更优选15-200g/L,进一步更优选17-180g/L的量提供。
示例性实施方案
下文描述示例性但非限制性的实施方案以进一步定义本发明。
一种由式L-(2)的L-草铵膦C
其中式L-(2)中的R
步骤1:将Ca(OH)
步骤2:将(NH
步骤3:通过从清澈滤液除去水而分离所需铵盐L-(1)。
如上所述的方法,其中步骤1中的蒸馏在大于70℃至130℃,优选75-120℃,尤其是80-110℃的温度下进行。就此而言,该蒸馏优选在大气压力(也称为常压)下进行。该蒸馏还可以在减压,优选100毫巴至低于大气压力,更优选100毫巴至小于1000毫巴,尤其是100-900毫巴下进行。就此而言,该蒸馏可以在20-110℃或30-120℃的温度下进行。
如上所述的方法,其中步骤2进一步包括加入醇,优选选自甲醇、乙醇、丙醇、异丙醇、丁醇及其混合物的醇,尤其是甲醇。就此而言,该醇优选在25-75℃,更优选30-70℃,甚至更优选35-65℃,尤其是40-60℃的温度下加入。
如上所述的方法,其中步骤2在步骤1之后在没有过滤步骤下进行。
如上所述的方法,其中式L-(2)的L-草铵膦烷基铵盐通过在两步法中使式DL-(1)的草铵膦铵盐去外消旋化而得到:
其中在第一步中使D-草铵膦氧化脱氨成PPO(2-氧代-4-(羟基(甲基)膦酰基)丁酸)用D-氨基酸氧化酶(DAAO)进行,以及在第二步中使用来自一种或多种胺供体的胺基团由转氨酶(TA)将该PPO胺化成式L-(2)的L-草铵膦C
如上所述的方法,其中式L-(2)的L-草铵膦C
如上文所述的方法,其中式L-(2)的L-草铵膦C
如上所述的方法,其中该DAAO酶选自来自圆红冬孢酵母(Rhodosporidiumtoruloides)(UniProt P80324)、变异三角酵母(Trigonopsis variabilis)(UniProtQ99042)、洁丽新韧伞(Neolentinus lepideus)(KZT28066.1)、里氏木霉(Trichodermareesei)(XP_006968548.1)或油脂丝孢酵母(Trichosporon oleaginosus)(KLT40252.1)的酶。
优选该DAAO酶是突变DAAO,更优选该突变DAAO是基于来自圆红冬孢酵母(Rhodosporidium toruloides)的序列的突变DAAO。
如上所述的方法,其中该TA酶是ω-转氨酶。
优选该TA酶是选自如下的S-选择性ω-转氨酶:节杆菌属(Arthrobacter sp.),巨大芽孢杆菌(Bacillus megaterium),肺炎克雷伯菌(Klebsiella pneumoniae)JS2F(S),苏云金芽孢杆菌(Bacillus thuringiensis)JS64(S),河流弧菌(V.fluvialis)JS17(S),假单胞菌属(Pseudomonas sp)KNK425(S),反硝化产碱菌(Alcaligenes denitrificans)Y2k-2(S),中间根瘤菌属(Mesorhizobium sp.)LUK(S),巨大芽孢杆菌(Bacillus megaterium)SC6394(S),腔隙莫拉氏菌(Moraxella lacunata)WZ34(S),大地双面神菌(Janibacterterrae)DSM13953(S),菊苣假单胞菌(Pseudomonas cichorii)DSM 50259(S),荧光假单胞菌(Pseudomonas fluorescens)ATCC49838(S),荧光假单胞菌(Pseudomonas fluorescens)KNK08-18(S),假单胞菌属(Pseudomonas sp)ACC(S),恶臭假单孢菌(Pseudomonas putida)NBRC14164(S),耐盐芽孢杆菌(Bacillus halotolerans)(S),枯草芽孢杆菌堆肥亚种(Bacillus subtilis subsp.stercoris)(S),枯草芽孢杆菌沙漠亚种(Bacillus subtilissubsp.inaquosorum)(S),植物内生芽孢杆菌(Bacillus endophyticus)(S),放射型根瘤菌(Rhizobium radiobacter)(S),青紫色素杆菌(Chromobacterium violaceum)DSM30191(S),铜绿假单胞菌(Pseudomonas aeruginosa)(S)Ingram等人(2007),柠檬节杆菌(Arthrobacter citreus)(S),新月柄杆菌(Caulobacter crescentus)(S),球形红杆菌(Rhodobacter sphaeroides)(S),脱氮副球菌(Paracoccus denitrificans)(S),极地单胞菌属(Polaromonas sp.)JS666(S),人苍白杆菌(Ochrobactrum anthropi)(S),鲍曼不动杆菌(Acinetobacter baumannii)(S),巴氏醋杆菌(Acetobacter pasteurianus)(S),越南伯克霍尔德氏菌(Burkholderia vietnamensis)(S),高嗜盐菌(Halomonas elongata)(S),草根围伯克霍尔德氏菌(Burkholderia graminis)(S),玫瑰红嗜热菌(Thermomicrobiumroseum)(S),嗜热球形杆菌(Sphaerobacter thermophilus)(S),热脱氮地芽孢杆菌(Geobacillus thermodenitrificans)(S),巨大芽孢杆菌(Bacillus megaterium)(S),蕈状芽孢杆菌(Bacillus mycoides)(S),嗜盐单胞菌(Halomonas sp.)CSM-2(S),红螺菌科细菌(Rhodospirillaceae bacterium)(S),拉布伦茨氏菌属(Labrenzia sp.)LAB(S),阿菲波菌属(Afipia sp.)P52-10(S),南海大洋棒菌(Oceanibaculum indicum)(S),球菌伊卢马托菌(Ilumatobacter coccineus)(S),贪食菌属(Variovorax sp.)KK3(S),卡瑞苯西思副伯克霍尔德氏菌(Paraburkholderia caribensis)(S),帕氏氢噬胞菌(Hydrogenophagapalleronii)(S),土壤红杆菌(Solirubrobacter soli)(S),动孢囊菌属(Kineosporiasp.)R_H_3(S),沙漠玫瑰单胞菌(Roseomonas deserti)(S),苜蓿中华根瘤菌(Sinorhizobiummeliloti)(S),羽扇豆博斯氏菌(Bosea lupine)(S),Bosea vaviloviae(S),中型假嗜酸菌(Pseudacidovorax intermedius)(S),伯克霍尔德氏菌属(Burkholderia sp.)UYPR1.413(S),大肠杆菌(Escherichia coli)K12(S),恶臭假单孢菌(Pseudomonas putida)(S),荧光假单胞菌(Pseudomonas fluorescens)(S),绿针假单胞菌(Pseudomonas chlororaphis)(S),波美罗伊硅杆菌(Silicibacter pomeroyi)(S),球形红杆菌(Rhodobacter sphaeroides)KD131(S),鲁杰氏菌属(Ruegeria sp.)TM1040(S),百脉根中慢生根瘤菌(Mesorhizobium loti)MAFF30399(S)或炭疽芽孢杆菌(Bacillusanthracis)(S)。
更优选该TA酶是突变ω-转氨酶。
最优选该TA是基于来自节杆菌属(Arthrobacter sp.)或巨大芽孢杆菌(Bacillusmegaterium)的序列的突变ω-转氨酶。
优选选择的ω-转氨酶突变体尤其描述于WO 2020/025577中。
一种通过使式DL-(1)的草铵膦铵盐去外消旋化而得到式L-(1)的L-草铵膦铵盐的方法,其中在起始步骤中使D-草铵膦氧化脱氨成PPO(2-氧代-4-(羟基(甲基)膦酰基)丁酸)用D-氨基酸氧化酶(DAAO)进行,并且在随后步骤中使用来自选自乙胺、正丙胺、异丙胺、正丁胺、仲丁胺、戊胺、仲戊胺、正己胺和仲己胺的胺供体的胺基团由转氨酶(TA)将该PPO胺化成式L-(2)的L-草铵膦C
其中式L-(2)中的R
以及在另一随后步骤中将(NH
以及其中在最后步骤中通过从该清澈滤液除去水而得到所需式L-(1)的L-草铵膦铵盐。
如上文所述的方法,其中所有方法步骤作为一锅法在单一容器中连续进行。
如上文所述的方法,其中各方法步骤作为多锅法在分开的容器中连续进行。
如上所述的方法,其中蒸馏步骤在大于70℃至130℃,优选75-120℃,尤其是80-110℃的温度下进行。就此而言,该蒸馏优选在大气压力(也称为常压)下进行。该蒸馏还可以在减压,优选100毫巴至低于大气压力,更优选100毫巴至小于1000毫巴,尤其是100-900毫巴下进行。就此而言,该蒸馏可以在20-110℃或30-120℃的温度下进行。
如上所述的方法,其中当加入(NH
如上所述的方法,其中该(NH
一种得到具有为任选包含一个或多个杂原子的(杂)芳族或(杂)脂族、(杂)环状或直链/支化开链的饱和或不饱和结构部分的有机结构部分R的式(6)的有机羧酸铵盐RCOONH
(A)第一步,其中通过加入Ca(OH)
(B)在第二步中通过加入硫酸铵置换式(5)的羧酸钙盐的钙离子并且随后通过过滤除去沉淀的石膏CaSO
如上文所述的方法,其中该中间体式(5)的羧酸钙由式(4)的羧酸的仲C
如上文所述的方法,其中步骤(A)中的蒸馏在大于70℃至130℃,优选75-120℃,尤其是80-110℃的温度下进行。就此而言,该蒸馏优选在大气压力(也称为常压)下进行。该蒸馏还可以在减压,优选100毫巴至低于大气压力,更优选100毫巴至小于1000毫巴,尤其是100-900毫巴下进行。就此而言,该蒸馏可以在20-110℃或30-120℃的温度下进行。
如上文所述的方法,其中步骤(B)进一步包括加入醇,优选选自甲醇、乙醇、丙醇、异丙醇、丁醇及其混合物的醇,尤其是甲醇。就此而言,该醇优选在25-75℃,更优选30-70℃,甚至更优选35-65℃,尤其是40-60℃的温度下加入。
如上文所述的方法,其中步骤(B)在步骤(A)之后在没有过滤步骤下进行。
制备方法
如上所述,本发明制备L-草铵膦方法涉及一种几步法,该方法的特征在于在第一步中使用含钙强碱如Ca(OH)
硫酸铵在该水溶液中离解,硫酸根离子形成沉淀的水不溶性硫酸钙(CaSO
方案2:
沉淀的石膏(或熟石膏)可以通过过滤容易地从该溶液除去,并且可以通过从含有铵盐L-(1)的剩余水溶液中除去该溶剂而得到纯净盐。
在本发明的优选实施方案中,如方案2a所示,优选的胺供体是异丙胺,其沸点为31.4℃并且因此可以容易地从该水溶液中除去。
方案2a:
将该新的本发明工艺步骤嵌入D,L-草铵膦的去外消旋化方法中,其由上述使D-草铵膦氧化脱氨成PPO开始。尽管在随后的氨基转移反应步骤之后可以由PPO直接得到L-草铵膦,但也得到显著量的副产物,即式L-(2a)的L-草铵膦异丙铵盐,其随后应如上文所述根据本发明进一步加工。
尽管如上文所述将本发明方法用于以及应用于制备L-草铵膦,但该转化是普遍适用的并且并不具体限于草铵膦或磷化合物,而是如下面的方案X所示还可以容易地应用于获得式(6)的羧酸铵盐,其中R为任选包含一个或多个杂原子的任何有机(杂)芳族或(杂)脂族、(杂)环状或直链/支化开链的饱和或不饱和结构部分。这里同样通过加入Ca(OH)
方案X:
方案X中的R可以是任何有机结构部分并且式(4)中的R
出于单独说明和表示的目的,下文在方案Xa中给出制备实施例,其中该盐为2-苯基丙酸异丙铵盐(7),这意味着阴离子是非磷羧酸根,在加入Ca(OH)
方案Xa:
该方法可以以“一锅法”进行或者在多个容器中,即以分开的转化进行。
若该反应在单一容器或器皿中进行,则该TA酶可以与该DAAO酶一起加入或者可以稍后加入,例如在已经允许该DAAO酶催化一些或基本所有该氧化脱氨之后加入。
这些酶可以通过许多方法加入该反应中。
一种方法是在微生物如大肠杆菌(E.coli)、酿酒酵母(S.cerevisiae)、巴斯德毕赤酵母(P.pastoris)等中表达该酶并且将全细胞作为全细胞生物催化剂加入该反应中。另一方法是表达该酶,裂解微生物并加入细胞裂解物。再一方法是从裂解物中纯化或部分纯化酶并将纯净或部分纯净的酶加入该反应中。可以与上述方法结合的另一方法是将酶固定于载体(示例性策略如Datta等人(2013)3Biotech.Feb;3(1):1-9所述)。若反应要求多种酶,则可以在一种或几种微生物中表达酶,包括在单一微生物中表达所有酶。
若反应要求多种酶,则可以在一种或几种微生物中表达酶,包括在单一微生物中表达所有酶。
可以与上述方法结合的另一方法是将酶固定于载体(示例性策略如Datta等人(2013)3Biotech.Feb;3(1):1-9所述)。如Datta等人所述且并不意欲是限制性的,酶单独或组合可以例如吸附于或者共价或非共价连接于天然或合成聚合物或无机载体(包括酶本身的聚集体)或者包埋于其中。一旦固定,则可以将酶和载体分散于本体溶液(bulksolution)中或者填充于床、柱或任何数目的类似方法以使反应溶液与酶相互作用。由于充气对于这里设想的DAAO反应是重要的,可以将泡罩塔或类似装置用于酶固定。作为实例,可以使反应混合物流过固定化酶的塔(流动反应),加入固定化酶的固定床或塔中,允许其反应并且从反应容器底部或顶部取出(活塞流),或者将其加入分散的固定化酶中并允许其反应,然后通过过滤、离心或类似方法(分批)除去固定化酶。因此,任何固定酶的方法可以用于本发明方法中。
该DAAO、TA和/或其他反应可以在缓冲剂中发生。
常用于生物转化反应中的示例性缓冲剂包括Tris缓冲剂、磷酸盐缓冲剂或任何Good缓冲剂,如2-(N-吗啉代)乙磺酸(MES);N-(2-乙酰胺基)亚氨基二乙酸(ADA);哌嗪-N,N'-二(2-乙磺酸)(PIPES);N-(2-乙酰胺基)-2-氨基乙磺酸(ACES);β-羟基-4-吗啉丙磺酸(MOPSO);氯化胆胺;3-(N-吗啉代)丙磺酸(MOPS);N,N-二(2-羟基乙基)-2-氨基乙磺酸(BES);2-[[1,3-二羟基-2-(羟甲基)丙-2-基]氨基]乙磺酸(TES);4-(2-羟基乙基)-1-哌嗪乙磺酸(HEPES);3-(二(2-羟基乙基)氨基)-2-羟基丙烷-1-磺酸(DIPSO);乙酰胺基甘氨酸,3-(N-三(羟甲基)甲基氨基)-2-羟基丙磺酸(TAPSO);哌嗪-N,N'-二(2-羟基丙磺酸)(POPSO);4-(2-羟基乙基)哌嗪-1-(2-羟基丙磺酸)(HEPPSO);3-[4-(2-羟基乙基)-1-哌嗪基]丙磺酸(HEPPS);三(羟甲基)甲基甘氨酸;甘氨酰胺;N,N-二羟乙基甘氨酸;或3-[[1,3-二羟基-2-(羟甲基)丙-2-基]氨基]丙烷-1-磺酸(TAPS)。额外的示例性缓冲剂配方可以在Whittall,J.和Sutton,P.W.(编辑)(2012)Front Matter,Practical Methods forBiocatalysis and Biotransformations 2,John Wiley&Sons,Ltd,Chichester,UK中找到。铵也可以用作缓冲剂。
该DAAO、TA和/或其他反应可以在不加入缓冲剂或者加入低水平(小于1mM)的缓冲剂(除了由于加入外消旋草铵膦铵盐而存在的铵之外)下发生。固定化DAAO和TA尤其在小于1mM磷酸盐缓冲剂存在下以及除了由于加入外消旋草铵膦铵盐而存在的铵外没有其他缓冲剂下可以是稳定且具有活性的。
在一些实施方案中,该反应在限定的pH范围内发生,该pH范围可以在pH 4-pH 10(例如pH 6-pH 9,如大约pH 7.5-pH 8)之间。
在一些实施方案中,该反应在限定的温度下发生。该温度可以保持在室温和溶剂沸点之间的点,最常见的是室温和50℃之间的点。
如所示那样,本文所述方法提供了基本纯净的L-草铵膦(而不是L-草铵膦和D-草铵膦的外消旋混合物)的组合物。基本纯净的L-草铵膦是指大于约80%,大于约85%,大于约90%,大于约95%,大于约96%,大于约97%,大于约98%或大于约99%的该D-草铵膦已经转化为L-草铵膦,得到一种与存在于该组合物中的D-草铵膦和L-草铵膦之和相比具有大于约80%,大于约85%,大于约90%,大于约95%,大于约96%,大于约97%,大于约98%或大于约99% L-草铵膦的组合物。就此而言,应理解的是术语“大于”是指至多100%的范围。
任选地,该L-草铵膦可以从该生物转化反应混合物中部分或完全提纯。可以使用许多替换程序,包括但不限于:a)使用合适的溶剂如醚或水在-20℃至该溶液的沸点之间变化的温度下由该生物转化混合物结晶,b)真空浓缩该生物转化混合物,然后使用合适的溶剂如醚或水在-20℃至该溶液的沸点之间变化的温度下由如此生产的浓缩物结晶,或者c)真空浓缩该生物转化混合物,然后在固定相如硅胶或十八烷基硅烷配体上分级提纯如此生产的浓缩物,后者作为在溶剂如DCM、甲醇(MeOH)、EtOAc、氨(NH
在一个实施方案中,该L-草铵膦不从该生物转化混合物分离并且得到一种包含D-草铵膦、PPO和L-草铵膦的组合物。该组合物可以直接用作除草组合物。
应理解的是本文所用术语仅是为了描述特定实施方案的目的,并且该术语意欲是说明性的而不是限制性的。除非另有定义,本文使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。在不背离本发明范围或要旨的情况下,本文所述和所示单独实施方案各自具有可以容易地与其他几种实施方案中任一种的特征分开或组合的离散组分或特征。任何所述方法可以以所述事件的顺序或者以逻辑上可能的任何其他顺序进行。尽管类似或等同于本文所述那些的任何方法和材料也可以用于本发明的实施或测试,现在描述代表性的示例方法和材料。
提供下列实施例作为说明而不是作为限制。
实施例
附图
本发明制备方法在获得羧酸的所需对映体的铵盐上的有效性在本文所示的下列图中证明:
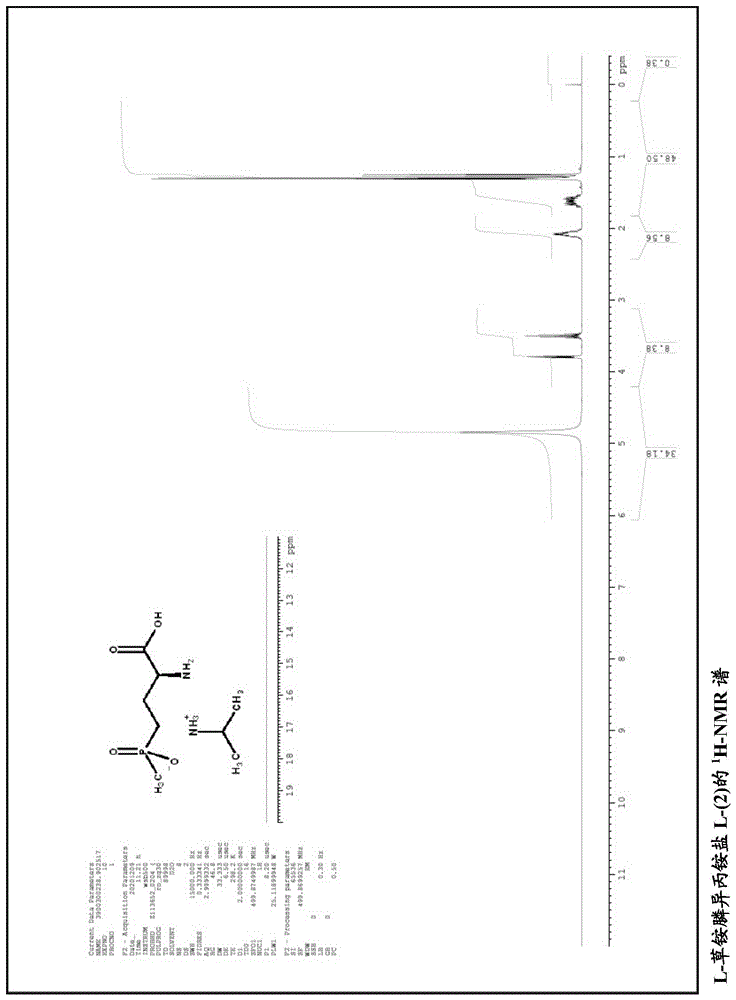
图1:L-草铵膦异丙铵盐L-(2)的
图2:L-草铵膦铵盐L-(1)的
图3:2-苯基丙酸异丙铵(7)的
图4:2-苯基丙酸铵(9)的
实施例1
使L-草铵膦异丙铵盐L-(2)转化为L-草铵膦铵盐L-(1):
在室温下将5.5g(30mmol)L-膦丝菌素L-(10)(Zeiss,H.-J.;J.Org.Chem.1991,56,1783)溶于80ml水和1.9g(33mmol)异丙胺中。将该溶液在室温下搅拌1小时,将0.5ml该清澈溶液的样品真空浓缩并进行H-NMR分析。所得固体残余物的
向该剩余溶液中加入75ml水和1.48g(20mmol)氢氧化钙。得到钙盐L-(3)的轻微浑浊溶液。在常压下通过蒸馏从该溶液除去100ml水。开始时沸点为88-95℃,随后接近蒸馏结束为100℃。通过用1N H
将钙盐L-(3)的剩余溶液冷却至50℃并用50ml甲醇进一步稀释。将2.64g(20mmol)(NH
实施例2
使2-苯基丙酸L-异丙基铵盐(7)转化为2-苯基丙酸铵(9):
在室温下将4.5g(30mmol)2-苯基丙酸和1.9g(33mmol)异丙胺溶于80ml水中并将该溶液在室温下搅拌1小时。取0.5ml所得清澈溶液的样品,真空浓缩并进行H-NMR分析。该固体残余物的
将该溶液用75mL水稀释并加入1.20g(16mmol)氢氧化钙。通过在常压下蒸馏从钙盐(8)的该悬浮液除去60ml水。开始时沸点为88-95℃,然后接近蒸馏结束为100℃。用1NH
将钙盐(8)的剩余悬浮液冷却至50℃并用50ml甲醇稀释。将2.10g(16mmol)(NH
- 一种用离子交换树脂分离L-草铵膦和葡萄糖酸的方法
- L-草铵膦中间体及L-草铵膦的制备方法
- L-草铵膦酸或L-草铵膦羧酸盐水溶液的制备方法